
Case Report
Austin J Clin Case Rep. 2014;1(9): 1044.
Burkitt Lymphoma: Thorax to Pelvis
S.K. Verma*, Neha
Department of pulmonary Medicine, KGMU, Lucknow 226003, India
*Corresponding author: S.K. Verma, Department of pulmonary Medicine, KGMU, Lucknow 226003, India
Received: August 04, 2014; Accepted: September 03, 2014; Published: September 05, 2014
Abstract
Burkitt lymphoma is a subgroup of non-Hodgkin’s lymphoma of high-grade which is aggressive and is composed of diffuse, undifferentiated malignant cells of lymphoid origin which are small and noncleaved. Dennis Burkitt first described this entity in 1956 in equatorial Africa. In humans, one of the fastest growing cancers is Burkitt lymphoma (BL), growth fraction close to 100%. Clinical course in Burkitt lymphoma is aggressive and rapid. It commonly occurs in children and young adults, with frequent involvement of bone marrow and central nervous system (CNS). These are considered to be medical emergencies and require immediate diagnostic and therapeutic intervention. Here we are reporting a case of Burkitt lymphoma with unusual presentation with involvement of both thorax and whole of the abdomen.
Keywords: Burkitt Lymphoma; Thorax; Pelvis; Children
Introduction
In Indian children, Burkitt lymphoma; accounting for 0.76% of solid malignant tumors. It is very rare malignancy. Most of the cases reported in literature presented either as jaw tumors or intra-abdominal malignancy and to the best of our knowledge, there is no Burkitt lymphoma case mimicking ours which presented as mass extending from thorax into abdomen and pelvis in continuity and hence we are reporting this case.
Case Presentation
A 16-year old non smoker male, presented to our outpatient department, with chief complaints of fever for 3 months, progressive breathlessness and left sided chest pain since 2 months. Fever was low grade, continuous, relieved on taking medications. Breathlessness was insidious in onset, gradually progressive initially MMRC grade 1 later on MMRC grade 4, increased on lying in right lateral position. Chest pain was insidious in onset, gradually progressive, localized to left side of hemi thorax, non-radiating, no diurnal or postural variation. Patient also complained of abdominal distention since 1 month which was progressive in nature and was associated with dull aching pain. There was no history of cough, hemoptysis, pedal edema, altered bowel habits, recurrent vomiting and reflux. On initial examination, patient was having a respiratory rate of 30/min and oxygen saturation of 92%. His Blood Pressure was 112/80 mmHg and pulse rate was 110/min. On general examination, pallor was present. Left axillary (4cmx3cm, single, mobile, firm) and left supraclavicular (2cmx1cm, single, mobile and firm) lymph nodes were palpable. On Respiratory System Examination, chest was asymmetrical with fullness on left hemi thorax, dilated veins present on left side of the chest, movement diminished on left side. Trail sign was present with right sided tracheal shift, apex beat was palpable in right 5th inter costal space medial to mid clavicular line. On percussion dull note was present over left hemi thorax. On auscultation, breath sounds diminished on left hemi thorax with decreased vocal resonance on left side. Per abdominal examination revealed tense and tender abdomen. Evaluation of rest of the systems was within normal limits. X ray chest PA view revealed left sided homogenous opacity with shift of trachea as well as mediastinum towards right side (Figure 1). USG whole abdomen was suggestive of a heterogenous predominantly hypoechoic lesion with internal vascularity covering whole abdomen extending from epigastrium mainly on left side to pelvis pushing the intra-abdominal vessels to right and bowel loops posteriorly with mild peritoneal collection. CT scan Thorax and abdomen was done which was suggestive of large ill defined heterogeneously enhancing soft tissue attenuation lesion extending from left hemithorax into abdominal cavity up to pelvis? Malignant (Figure 2,3,4). FNAC of left axillary lymph node showed population of abnormal cells with dual population, majority being large cells with moderate amount of cytoplasm with vacuolation. Nuclear chromatin is coarse, majority of cells showed round nuclear outline. Cytomorphology was suggestive of non Hodgkin’s lymphoma (Burkitt type) (Figure 5,6). Bone Marrow examination revealed early lymphomatous infiltration. However, the patient’s attendants denied further workup and treatment.
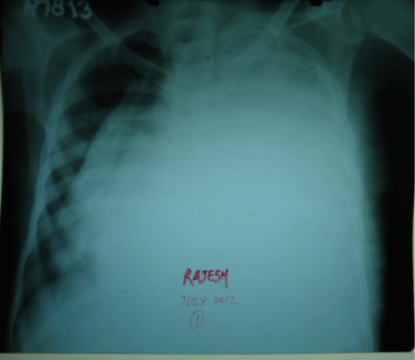
Figure 1: Chest X Ray PA view showing left sided homogenous opacity with tracheal and mediastinal shift towards right side.
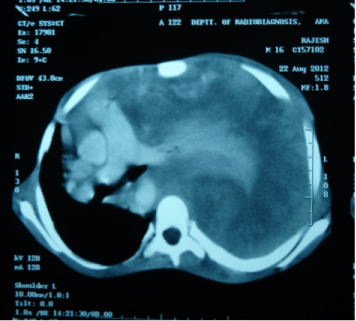
Figure 2: CT Scan Thorax suggestive of left sided intrathoracic mass with mediastinal shift.
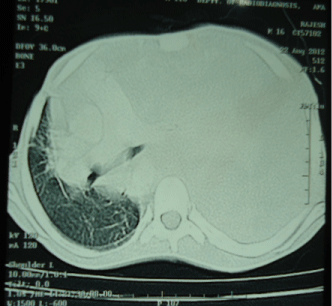
Figure 3: CT Scan Thorax suggestive of left sided intrathoracic mass with mediastinal shift.
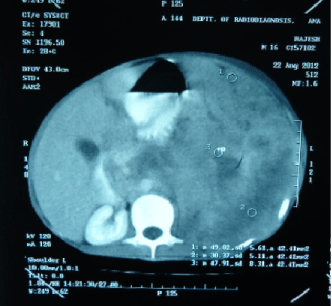
Figure 4: CT Scan abdomen showing left sided intra-abdominal mass shifting structures toward right.
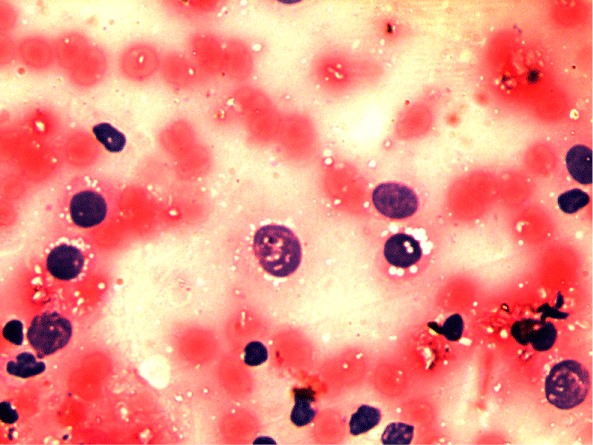
Figure 5: Cytomorphology suggestive of NON HODGKINS LYMPHOMA (BUKITT TYPE).
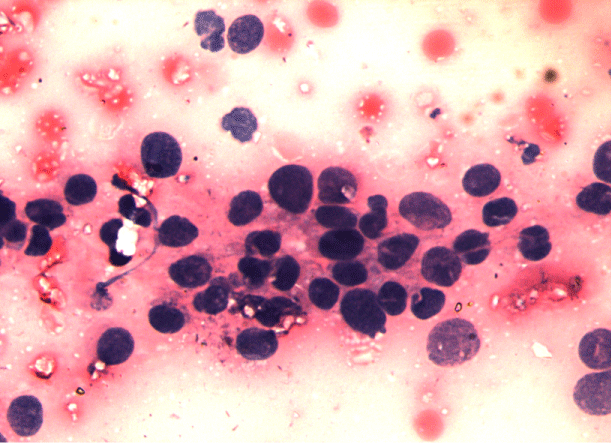
Figure 6: Cytomorphology suggestive of NON HODGKINS LYMPHOMA (BUKITT TYPE).
Discussion
Burkitt lymphoma, one of the highly aggressive B-cell non- Hodgkin lymphomas, is characterized by the translocation and deregulation of the c-myc gene on chromosome 8 [1]. With a doubling time of less than 24 h, it is the fastest growing human tumor [2,3]. Burkitt lymphoma can be divided into three main clinical variants: 1. the endemic, 2. the sporadic and the 3. Immunodeficiency associated variants. The endemic variant is commonly found in equatorial Africa. It is the most common malignancy among children in that area. Reduced resistance to Epstein Barr virus (EBV) was attributed to the coexistence of chronic malaria in these children. The common sites involved include the jaw among other facial bones, distal ileum, cecum, ovaries, kidney or the breast. The sporadic variant of BL is seen outside of Africa. The tumor cells show a similar appearance to that of classical or endemic Burkitt lymphoma. Impaired immunity is once again recognized to favor infection with EBV. The ileocecal region is commonly involved. Jaw involvement is less common when compared to the endemic variant. Immunodeficiency-associated BL is usually associated with HIV infection [4] or occurs in post-transplant patients taking immunosuppressive medications. Mutation and over expression of the c-myc oncogene is a characteristic feature. It results from a translocation between chromosomes 8 and 14, t (8;14) (q34;q32). Other translocations reported to cause c-myc over expression include t (2;8) (p12;q24) and t(8;22) (q24;q11). BL occupies 2% of cancers of the same blood cells [5]. Clinical manifestations of BL depend on the primary and secondary site of involvement. It commonly presents as an abdominal (sporadic type) or head and neck (endemic type) disease with involvement of bone marrow or CNS. Manifestations include painless rapidly growing lymph node swelling, cough, superior vena cava syndrome, dyspnea with thoracic extension, abdominal masses, intestinal obstruction, intussusceptions like symptoms and ascites or localized bony pain. Burkitt’s lymphoma is reported to present as abdominal or pelvic mass in 45% cases and in the gastrointestinal tract involvement per se in 22.5% [6]. Contiguous involvement of thorax, abdomen and pelvis is rare. Our patient presented with dyspnea, chest pain, fever and progressive abdominal distension at the same time probably due to the contiguous involvement of thorax, abdomen and pelvis. A separate staging system for BL has been developed by Ziegler (1981), whereas Levine et al. (1982), classified the cases of American BL as follows: [7].
Stage I: single tumor mass (extra-abdominal 1A or abdominal 2A).
Stage II: two separate tumor masses on the same side of the diaphragm.
Stage III: involvement of more than two separate masses or disease on both the sides of the diaphragm. Stage IV: pleural effusion, ascitis or involvement of the central nervous system (malignant cells in the cerebrospinal fluid) or bone marrow.
No racial predilection is reported and males are affected 2-3 times more often than females. Histologically, Burkitt’s cells are homogeneous in size and shape, with round to oval nuclei and slightly coarse chromatin, with multiple nucleoli and intensely basophilic vacuolated cytoplasm containing contains neutral fat. Immunohistochemistry by staining with Ki-67, CD-19, CD-20, CD- 22, CD-79a protein may be useful for diagnosis [8,9]. The hallmark of Burkitt lymphoma (BL) is the presence of a “starry sky” appearance (also observed in other highly proliferative lymphomas), caused by the presence of scattered macrophages phagocytizing cell debris and apoptotic cells. The primary modality of treatment is multi-agent systemic chemotherapy along with intrathecal chemotherapy. Debulking surgery is rarely useful and is used mainly to obtain tissue for biopsy and diagnosis. Radiation therapy is useful in the presence of CNS involvement in BL, acute SVC, and acute paraplegias. Regimens commonly used include COPAD (cyclophosphamide, vincristine, prednisone and doxorubicin) or COMP (cyclophosphamide, vincristine, methotrexate, 6-mercaptopurine and prednisone). Intrathecal Chemotherapy with intrathecal Methotrexate, hydrocortisone, or Ara-C is used in moderate to severe disease. Patients who develop progressive or relapsed disease require reinduction chemotherapy and either allogeneic or autologous stem cell transplantation. Current intensive chemotherapy regimens have shown long term disease free survival in about 90% of pediatric and 50-60% of adult patients with BL. Massive acute destruction of the tumor cells during initial chemotherapy due to the rapid growth rate may result in tumor lysis syndrome requiring renal dialysis. In the abdominal form of the disease, rapid tumor growth may result in intestinal obstruction; renal failure may occur from tumor infiltration of the kidneys. After extensive search of literature, we could not find out the similar type of case report so we can say that to the best of our knowledge our case appears to be first of its type.
References
- Ali H Kanbar. . In: DeVita VT Jr, Hellman S, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. 2. 5th edn. Philadelphia, Pa: Lippincott Williams 7a Wilkins. 1997; 2157-9.
- Ugar DA, Bozkaya S, Karaca I, Tokman B, Pinarli FG. Childhood craniofacial Burkitt's lymphoma presenting as maxillary swelling: report of a case and review of literature. J Dent Child (Chic). 2006; 73: 45-50.
- Wood RE. Malignant diseases of the jaws. In: White SC, Pharaoh MJ, editors. Oral radiology: Principles and interpretations. Mosby: St Louis. 2004. 450-451.
- Bellan C, Lazzi S, De Falco G, Nyongo A, Giordano A, Leoncini L, et al. Burkitt's lymphoma: new insights into molecular pathogenesis. J Clin Pathol. 2003; 56: 188-192.
- Turgeon, Mary Louise. Clinical hematology: theory and procedures. Hagerstown, MD: Lippincott Williams & Wilkins. ISBN 0-7817-5007-5. "Frequency of lymphoid neoplasms. (Source: Modified from WHO Blue Book on Tumour of Hematopoietic and Lymphoid Tissues.)" 2005; 283.
- Biko DM, Anupindi SA, Hernandez A, Kersun L, Bellah R. Childhood Burkitt lymphoma: abdominal and pelvic imaging findings. AJR Am J Roentgenol. 2009; 192: 1304-1315.
- Patton LL, McMillan CW, Webster WP. American Burkitt's lymphoma: a 10-year review and case study. Oral Surg Oral Med Oral Pathol. 1990; 69: 307-316.
- Pramanik R, Paral CC, Ghosh A. Pattern of solid malignant tumours in children--a ten-year study. J Indian Med Assoc. 1997; 95: 107-108, 115.
- Ferry JA. Burkitt's lymphoma: clinicopathologic features and differential diagnosis. Oncologist. 2006; 11: 375-383.