
Case Report
Austin J Clin Case Rep. 2014;1(11): 1052.
Epithelioid Angiomyolipoma – The Treatment Conundrum
Keller A1*, So A2 and Strutton G2
1Wesley Research Institute, Brisbane, Australia
2Department of Anatomical Pathology, Princess Alexandra Hospital, Australia
*Corresponding author: Keller A, Wesley Research Institute, Wesley Private Hospital, 451 Coronation Drive, Brisbane, QLD, 4066, Australia
Received: August 25, 2014; Accepted: September 25, 2014; Published: September 26, 2014
Abstract
A 21 year old female was transferred to our institution with generalised abdominal pain and an incidental finding of an exophytic 4cm enhancing upper pole renal mass on Computerised Tomography (CT). Her abdominal pain was subequently diagnosed as being secondary to constipation. The diagnosis of a likely Renal Cell Carcinoma (RCC) was explained to the patient and she was booked for a laprascopic mobilisation open partial nephrectomy. Histologic examination demonstrated adipocytes, spindle cells and abnormal vessels. The tumour tested postive for melanocytic and smooth muscle markers. Ten percent of the tumour consisted of epithelioid cells with scattered mitotic figures. The diagnosis was that of a fat poor angiomyolipoma (AML) with a small focus of epithelioid AML. Resection was clear of margins. We elected to surveil rather than electively add adjuvant therapies. At 12 month follow-up the patient remains disease free.
Keywords: AML; Epithelioid; Renal cell carcinoma; PEComa; Angiomyolipoma
Case Presentation
A 21 year old female was transferred to our institution with generalised abdominal pain and an incidental finding of an exophytic 4cm enhancing upper pole renal mass on Computerised Tomography (CT) (Figure 1,2). No focal stranding around the lesion or other abnormality was found other than moderate faecal loading.

Figure 1: Axial CT Non-Contrast enhanced image of an exophytic mass arising from the superior pole the left kidney. No fat density is visible within the mass.
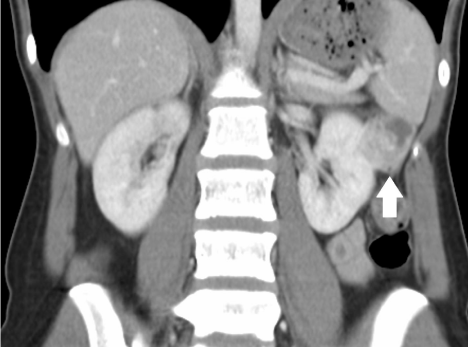
Figure 2: Coronal CT Intravenous Contrast enhanced image of renal mass demonstrating tissue enhancement.
Routine blood tests had demonstrated only a mild leukocytosis, which subsequently returned to normal levels on repeated blood tests the day following admission. Serum creatinine was normal. Urinalysis was unremarkable. Her abdominal pain was subsequently diagnosed as being secondary to constipation.
The diagnosis of a likely Renal Cell Carcinoma (RCC) was explained to the patient and she was booked for an elective open partial nephrectomy. The procedure was performed without complication. She had an uneventful recovery and was discharged 3 days following her operation.
Results
Histologic examination demonstrated adipocytes, spindle cells and abnormal vessels (Figure 3,4). The tumour tested positive for melanocytic and smooth muscle markers (Figure 5). 10% of the tumour consisted of epithelioid cells with scattered mitotic figures (Figure 6). The diagnosis was that of an Angiomyolipoma (AML) with a small focus of epithelioid AML. We considered adding elective adjuvant therapies, but due to the small size of the primary tumour, the presence of clear margins and the patient’s young age we elected for surveillance instead. At 12 month interval scanning she currently remains clear of recurrence. Post-operatively she had a mild rise in her serum creatinine, this has subsequently returned to her pre-operative baseline.

Figure 3: Low magnification image of region of “classic AML” with spindle cells and adipose tissue.

Figure 4: Higher magnification of normal spindle cells within tumour, demonstrating uniform nuclei and small amounts of cytoplasm.
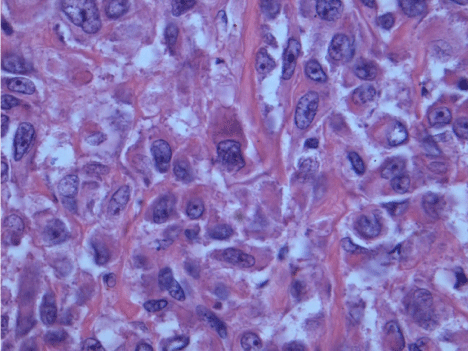
Figure 5: Positivity of melanocytic(HMB45 and Melan A) and smooth muscle markers(SMA and Desmin) confirming diagnosis of PEComa.
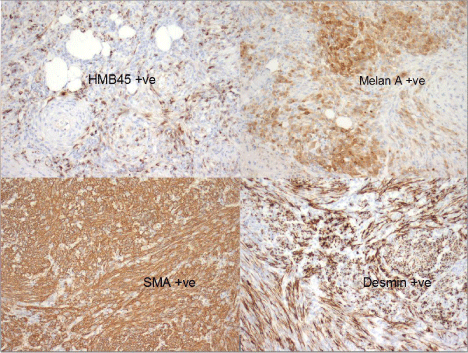
Figure 6: Higher magnification of focus of epithelioid cells. These cells demonstrate nuclear atypia and increased amounts of eosinophilic cytoplasm compared to spindle cells in “classic” AML.
Discussion
Renal angiomyolipoma is a mesenchymal tumour of the kidney. It is composed of blood vessels, smooth muscle and fat cells. AML has traditionally been described as a hamartoma, however, this is incorrect, it is a true clonal neoplasm. Angiomyolipomas belong to the greater classification of perivascular epithelioid cell tumours or PEComas [1,2]. PEComas include clear cell sugar tumour of the lung and lymphangiomyomatosis as well as AML [3]. PECs stain positive for melanocytic markers including HMB45 and melanin and smooth muscle markers such as actin and stain negative for epithelial markers [4].
PEComas are found in a wide distribution throughout the body including colon, pancreas, retroperitoneum, heart, eye, bone, nasopharynx, urinary bladder as well as the kidney [5]. Epithelioid AML is a rare subtype of PEComa. Approximately 120 cases have been described in the literature, [6] however; the true incidence is likely higher as in several retrospective studies it has often mistakenly been diagnosed histologically as classic AML or renal cell carcinoma [7]. It contrasts from classic AML by the presence of epithelioid cells and absence of vascular structures and adipocytes. Cells contain eosinophilic cytoplasm with hyperchromatic and pleomorphic nuclei. Intratumor necrosis and haemorrhage are more frequently seen than in classic AML [8].
Whilst most PEComas, including classical AML behave in a benign manner there is a growing body of evidence to suggest that epithelioid variants can behave aggressively with local recurrence, metastasis and death [9]. Case reports have described rapidly progressive metastatic disease and death within 3 months of diagnosis [10]. Other reports describe late local recurrence up to 9 years after initial nephrectomy. Most fatal cases have been in patients with primary tumours of 15 centimetres or greater. Lungs are the most frequent metastatic site with liver, with CNS and bone also common [6,11]. Pure epithelioid AML and epithleioid AML arising within classic AML appear to display similarly aggressive natural histories based on the few case reports [12,13].
PEComas in general are stongly associated with the Tuberous Sclerosis Complex (TSC). Tuberous sclerosis is a syndrome characterised by mental retardation, seizures and various cellular proliferations, including PEComatosis with an incidence of 55- 75% [14]. Tuberous sclerosis is caused by mutation of the tumour suppressor genes TSC1 or TSC2. TSC acts to suppress various oncogenes including mTOR. Loss of these genes has also been found in sporadic AMLs and PEComas [15]. Various agents targeting mTOR have been trialled in patients with AML with success. Trials of rapamycin and sirolimus have shown a 30-53% reduction in baseline tumour size at 12 months. Regrowth of tumour was seen at cessation of therapy almost universally, and in some series tumour returned to baseline size despite persistence of therapy [16-18].
Wolff et al have successfully treated local recurrence and metastasis of epithelioid AML with sirolimus and temsirolimus with success. Patients in his small series had both sporadic as well as tuberous sclerosis associated neoplasms. Follow-up was, however, short at less than 12 months and interstitial pneumonitis and opportunistic infections complicated treatment [19]. No large RCTs have been performed to evaluate the utility of mTOR inhibitors for the treatment of epithelioid AML.
Interferon has also been used in addition to surgical excision in one case of primary epithelioid PEComa of the bladder. The patient was free of recurrence 48 months after diagnosis [20].
Due to the rarity of this condition most scholarly articles are case reports or small case series with differing diagnostic criteria. The largest study looked at 41 cases of “pure” epithelioid AMLs, whereby the tumour contained solely epithelioid components with no adipocytes or vascular structures [6]. Of the 41 patients nine had tuberous sclerosis, the mean age at diagnosis was 40.7 years, male to female ratio was 1:1. Of the 41 cases, followup was available for 33 with mean followup of 44.5 months. Recurrence was seen in 17%, metastasis in 49% and 33% died of their disease. The authors have developed an algorithm for risk of disease progression. The cases were graded as demonstrating either carcinoma-like growth pattern A or plump spindle cells with diffuse growth, pattern B. Risk factors were: presence of tuberous sclerosis complex or concurrent classic AML, tumour size of >7cm, presence of necrosis, invasion of renal vein or peri-nephric fat and growth pattern A. Low risk tumours had 0-1 risk factor, intermediate 2-3 and high risk 4 or more. Disease progression was seen in 15% of low risk, 64% with intermediate risk and 100% of cases with high risk features.
Data on “nonpure” epithelioid AML is limited, and as such we are unable to determine whether described prognostic factors for “pure” epithelioid AML are directly attributable to our patient. Applying Folpe et al’s prognostic criteria to our patient would place her at low risk of disease progression. Due to her low risk stratification, young age, small volume of disease, and clear margins we are monitoring for recurrence with regular imaging, rather than electively adding adjuvant therapies. She has remained disease free at 12 months following her initial resection on follow-up scanning. There is no data currently to quantify a suitable surveillance period or interval.
References
- Eble JN. Angiomyolipoma of kidney. Semin Diagn Pathol. 1998; 15: 21-40.
- Folpe AL. Neoplasms with perivascular epithelioid cell differentiation (PEComas) World Health Organisation Classifcation of Tumours: Pathology of Genetics of Tumours of Soft Tissue and Bone. Fletcher CDM, Unni KK, Mertens F, Editor. IARC Press Lyon France. 2002; 221-222.
- Pea M, Bonetti F, Zamboni G, Martignoni G, Fiore-Donati L, Doglioni C. Clear cell tumor and angiomyolipoma. Am J Surg Pathol. 1991; 15: 199-202.
- Martignoni G, Pea M, Bonetti F et al. Renal epithelioidoxyphylic neoplasm (REON): a pleomorphic monophasic variant of angiomyolipoma. Int J Surg Pathol.1994; 2: 539.
- Bleeker JS, Quevedo JF, Folpe AL. "Malignant" perivascular epithelioid cell neoplasm: risk stratification and treatment strategies. Sarcoma. 2012; 2012: 541626.
- Nese N, Martignoni G, Fletcher CD, Gupta R, Pan CC, Kim H, et al. Pure epithelioid PEComas (so-called epithelioid angiomyolipoma) of the kidney: A clinicopathologic study of 41 cases: detailed assessment of morphology and risk stratification. Am J Surg Pathol. 2011; 35: 161-176.
- Varma S, Gupta S, Talwar J, Forte F, Dhar M. Renal epithelioid angiomyolipoma: a malignant disease. J Nephrol. 2011; 24: 18-22.
- Huang S, Chen Y, Wu C, et al. Epithelioid Angiomyolipoma of Kidneys. Acta Nephrologica. 2012; 26: 28-31.
- Chen J, Wang P, Wang CJ, Cai SL, Ren GP, Li YY. Highly aggressive epithelioid renal angiomyolipoma with a very poor prognosis. Chin Med J (Engl). 2010; 123: 765-766.
- Yamamoto T, Ito K, Suzuki K, Yamanaka H, Ebihara K, Sasaki A. Rapidly progressive malignant epithelioid angiomyolipoma of the kidney. J Urol. 2002; 168: 190-191.
- Folpe AL, Mentzel T, Lehr HA, Fisher C, Balzer BL, Weiss SW. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol. 2005; 29: 1558-1575.
- Park HK, Zhang S, Wong MK, Kim HL. Clinical presentation of epithelioid angiomyolipoma. Int J Urol. 2007; 14: 21-25.
- Martignoni G, Pea M, Rigaud G, Manfrin E, Colato C, Zamboni G, et al. Renal angiomyolipoma with epithelioid sarcomatous transformation and metastases: demonstration of the same genetic defects in the primary and metastatic lesions. Am J Surg Pathol. 2000; 24: 889-894.
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006; 355: 1345-1356.
- Kenerson H, Folpe AL, Takayama TK, Yeung RS. Activation of the mTOR pathway in sporadic angiomyolipomas and other perivascular epithelioid cell neoplasms. Hum Pathol. 2007; 38: 1361-1371.
- Cabrera López C, Martí T, Catalí V, Torres F, Mateu S, Ballarín Castán J, et al. Effects of rapamycin on angiomyolipomas in patients with tuberous sclerosis. Nefrologia. 2011; 31: 292-298.
- Dabora SL, Franz DN, Ashwal S, Sagalowsky A, DiMario FJ Jr, Miles D, et al. Multicenter phase 2 trial of sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors regress and VEGF- D levels decrease. PLoS One. 2011; 6: e23379.
- Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008; 358: 140-151.
- Wolff N, Kabbani W, Bradley T, Raj G, Watumull L, Brugarolas J. Sirolimus and temsirolimus for epithelioid angiomyolipoma. J Clin Oncol. 2010; 28: e65-68.
- Parfitt JR, Bella AJ, Wehrli BM, Izawa JI. Primary PEComa of the bladder treated with primary excision and adjuvant interferon-alpha immunotherapy: a case report. BMC Urol. 2006; 6: 20.