
Case Report
Austin J Clin Case Rep. 2023; 10(7): 1303.
A Childhood-Onset Nemaline Myopathy Caused by Novel Compound Heterozygote Variants in the Nebulin Gene
Jing Xu*; Jie Wu; Xiaowen Li; Wei Jiang; Chunyang wang
Department of Neurology, Tianjin Neurological Institute, Tianjin Medical University General Hospital, China
*Corresponding author: Jing Xu Department of Neurology, Tianjin Medical University General Hospital, 154 Anshan Road, Heping District, Tianjin, 300052, China. Email: jingxu01@tmu.edu.cn
Received: September 12, 2023 Accepted: October 07, 2023 Published: October 14, 2023
Abstract
Congenital myopathies are clinically and genetically heterogeneous disorders, which often remain genetically undiagnosed for many years. Here we present a 20-year-old patient showing gradually deteriorated proximal muscle weakness and rod-shaped structures found in muscle fibers was suspected of having nemaline myopathy. Whole-exome sequencing and subsequent Sanger sequence analysis for the patient revealed a pathogenic mutation in NEB gene c.21522+3 (IVS 144) A>G and c.23722 (exon 165) A>T. Based on genetic analyses, we identified two novel, compound- heterozygous variants in the NEB gene, which cause a childhood- onset nemaline myopathy.
Keywords: Congenital myopathy; Nemaline myopathy; NEB; Next Generation Sequencing
Introduction
Nemaline Myopathy (NM) is a hereditary muscle disorder with a wide range of severity. The most common clinical symptoms include early-onset muscle weakness of proximal limb, neck flexors, weakness of respiratory muscles [1]. NM was historically defined by the muscle biopsy finding of nemaline rods [2].
Mutations in 12 genes have been associated with NM. The most common mutations are NEB (encoding nebulin) [3,4] and ACTA1 (skeletal muscle a-actin) [5,6], other mutant genes including TPM2 (β-tropomyosin) [7], TPM3 (a-tropomyosin) [8,9], KBTBD13 (Kelch repeat and BTB domain-containing protein 13) [9], CFL2 (cofilin-2) [10], KLHL40 (Kelch-like family member 40, KLHL40) [11], KLHL41 (Kelch-like family member 41, KLHL41) [12], LMOD3 (leiomodin-3) [13], MYPN (myopalladin) [14], TNNT1 (troponin T1) [15,16] and TNNT3 (troponin T3) [17].
Homozygous or compound heterozygous mutation in the nebulin gene on chromosome 2q23 is responsible of NM [18]. NEB gene encodes nebulin, a giant cytoskeletal protein that plays a role in specifying and maintaining the length of actin thin filaments in striated muscle [19]. Here, we report a case of NM in a 20-year-old boy using next-generation sequencing.
Case Report
A 20-year-old male patient presented in the clinic complaining of a longstanding history of weakness since early childhood. He reported that he never gained the ability to run. Symptoms were progressive, so that he had difficulty in climbing stairs. He denied any history of muscle pain and stiffness. There was no history of diplopia, shortness of breath, or change in urine color. The weakness was not fluctuating and not associated with dysphagia or facial weakness. He has one sister and one brother, both of them died when they were infants with unknow cause. Both parents did not report neuromuscular symptoms and were normal upon clinical examination (Figure 1).
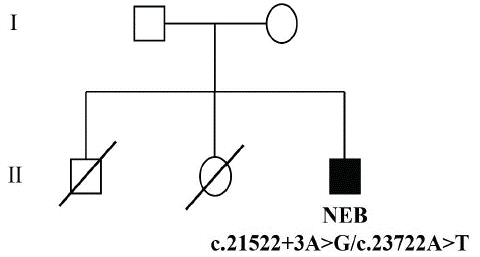
Figure 1: Results of DNA sequence. Pedigree symbols. Square: male; circle: female; unfilled: unaffected or unknown; filled: affected; a diagonal line through a symbol, death.
On examination, he had a symmetrical face with no ptosis, ophthalmoplegia, or dysmorphic characteristics. No muscle wasting or fasciculations in the tongue or extremities were observed. Inspection of skeletal muscle and testing of muscle strength showed atrophy and weakness of the proximal and distal leg muscles (hip extensors and knee flexors: 3/5; foot extensors: 4/5). There was no rigid spine, axial atrophy or scapula alata.
Serum creatine kinase levels and vitamin D levels were normal. Imaging of the spinal cord showed no abnormalities. Echocardiography and pulmonary function tests were normal. Electromyography revealed myopathic potentials with normal nerve conduction evaluation for right upper and lower extremities. A first skeletal muscle biopsy revealed typical nemaline rods in the modified Gomori-trichrome-stain (Figure 2).
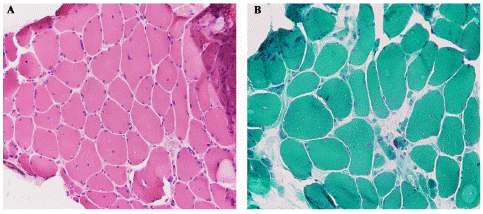
Figure 2: Left quadriceps femoris muscle biopsy from our patient by conventional microscopy. Hematoxylin and eosin (H&E) staining (×100) showed presence of abundant rods (dark pink substance), a wide variation in fiber size, as well as occasional atrophic small round fibers in certain parts (A). Modified Gomori trichrome staining (×100) reveals frequent subsarcolemmal dark nemaline rods which also aggregate in the intermyofibrillar spaces (B).
Whole-exome sequencing and subsequent Sanger sequence analysis for the patient revealed a pathogenic compound heterozygous variant in NEB gene c.21522+3 (IVS 144) A>G and c.23722 (exon 165) A>T (Table 1). Unfortunately, his parents didn't have genetic testing. He was diagnosed with autosomal recessive nemaline myopathy with two novel pathogenic variants in the NEB gene.
![]()
Gene
Site
Sequence variants
State
ACMG evidence
of pathogenicityNEB
Exon165
IVS144
c.23722A>T
(p.R7908*)
Chr2:152362712
c.21522+3A>G
-
chr2:152389953Het
Het
VUS
Likely Path
Het heterozygote, Likely Path likely pathogenic, VUS variant of uncertain significance, ACMG American College of Medical Genetics
Table 1: List of found variants in our patient.
Discussion
The NM constitute a large proportion of the congenital or structural myopathies. The causative genes are at least twelve, encoding structural or regulatory proteins of the thin filament, and the clinical picture as well as the histological appearance on muscle biopsy vary widely.
Among these known genes, mutations in the NEB gene are the most common cause of autosomal recessive NM, corresponding to around 50% of the cases [20]. This gene is huge, with 183 exons spanning 249 kb of the genomic sequence, and encodes the nebulin protein of approximately 600–900 kDa [19,21,22]. Therefore, the molecular study of this gene was a great challenge in previous years. We collected more than 80 NEB mutation pattern with literature review (Table 2). More recently, with the advance of next-generation sequencing methodologies, the screening of the NEB gene has been applied in larger cohorts of NM patients, and more mutations have been identified. Yin et al reported that NEB gene mutation was the most common mutation in a 16 NM patient cohort, in which splicing change c.21522 +3A > G is hotspot mutation in Chinese NM patients (33742414). Another cohort including 48 NM patients found that NEB was the most frequent causative gene in this Chinese cohort, followed by ACTA1. Notably, one NEB splicing mutation, c.21417+3A>G causing exon 144 splicing was found in 52.9% of NEB variant-carrying patients [23]. In Brazilian patients, the most common mutations are also NEB. Therefore, previous study suggested that considering the high frequency of NEB mutations and the complexity of this gene, NGS tools should be combined with CNV identification, especially in patients with a likely non-identified second mutation [24].
![]()
Mutation site
Ethnicity
References
A homozygous mutation in NEB intron 122
Lebanese
[30]
c.20131C > T; c.674C > T
Unknown
[33]
c.1999-2A>G; c.1999-2A>G
Unknown
[35]
c.6915+1G>T; c.14910+3G>C
Unknown
[36]
c.8501delA; c.1674+2T>C
Brazilian
[24]
c.24189_24192dup; c.20466+2T>A
Brazilian
[24]
c.24304_24305dup; c.24304_24305dup
Brazilian
[24]
c.8889+1G>A; c.6869_6870insTGC
Brazilian
[24]
c.22170G>A; c.24579G>C
Brazilian
[24]
c.24735_24736del; IVS122:c.191028_19102-4del
Brazilian
[24]
c.23601_23602del; c.2835+5G>C
Brazilian
[24]
c.23878_23881dup; c.25405-1G>C
Brazilian
[24]
c.1623delT; duplication of exons 82 to 105
Brazilian
[24]
c.3648del; del exon 29
Brazilian
[24]
c.5343+5G>A; c.5343+5G>A
Brazilian
[24]
c.21076C>T; c.24192_24193insTCAA
Brazilian
[24]
c.19944G>A; c.16423A>T
Brazilian
[24]
c.5343+5G>A; c.2943G>A
Brazilian
[24]
c.21522+119C>G; c.21522+119C>G
Algerian
[37]
c.23122-1G>C; c.21522+3A>G
Chinese
[34]
c.14837dupA; c.3758C>A
Chinese
[34]
c.21522+3A>G; c.11164C>T
Chinese
[34]
c.21522+3A>G; c.4417C>T
Chinese
[34]
c.21522+3A>G; c.23233-1G>T
Chinese
[34]
c.21522+3A>G; c.5343+1G>A
Chinese
[34]
c.21417+3A>G; c.20360_20361insA
Chinese
[23]
c.21793C>T; c.21417+3A>G
Chinese
[23]
c.21417+3A>G; c.5574_5575ins
Chinese
[23]
c.4189C>T; Exon 82 duplication
Chinese
[23]
c.1623delT; c.21417+3A>G
Chinese
[23]
c.21417+3A>G; c.19211delT
Chinese
[23]
c.13669C>T; c.2311-2A>C
Chinese
[23]
c.19944G>A; c.6029del
Chinese
[23,38]
c.7818delG; c.24579G>A
Chinese
[23,38]
c.17367G>A; c.21417+3A>G
Chinese
[23]
c.21417+3A>G; c.12019-10G>A
Chinese
[23]
c.21417+3A>G; c.18917G>A
Chinese
[23]
c.21605G>C; c.5789A>T; c.613A>T
Chinese
[23]
c.21417+3A>G; c.8479C>T
Chinese
[23]
c.21417+3A>G; c.1263dupA
Chinese
[23]
c.4352delC; c.1470+5G>T
Chinese
[23]
c.21417+3A>G; c.16465A>G
Chinese
[23]
c.24209_24212dupTGTT; c.183_184ins
Chinese
[23]
c.7212T>G; c.21417+3A>G
Chinese
[23]
c.18187C>T; c.21417+3A>G
Chinese
[23]
c.6195dupG; c.21417+3A>G
Chinese
[23]
c.8394T>G; c.21417+3A>G
Chinese
[23]
c.5924C>T; c.10976A>G; c.24650G>A
Chinese
[23]
c.21417+3A>G; c.36G>T
Chinese
[23]
c.22037A>T; c.21417+3A>G
Chinese
[23]
c.24314_24317dupTGTT; c.19732-3C>A
Chinese
[23]
c.21417+3A>G; c.19751T>G
Chinese
[23]
c.15187dupC; c.8350G>A
Chinese
[23]
c.21485A>C; c.8434C>T; c.2017T>C
Chinese
[23]
c.24375_24378del; c.18340delA
Chinese
[23]
c.23246_23249delAGTA; c.24182_24185dupAACA
Chinese
[23]
c.7653C>G
Chinese
[23]
c.2859T>G
Chinese
[23]
c.20131C>T
Chinese
[23]
c.3567 + 1G>A; c.6734dupA
Chinese
[38]
c.3255+1G>T; c.7165delA
Chinese
[39]
c.24372_24375dup
Caucasian
[28]
c.2310+5G > A; c.17779_17780delTA
Unknown
[40]
c.19653G>A; c.25441C>T
Unknown
[41]
c.18676C>T; c.9812C>A
Chinese
[42]
c.20131C>T; c.9046C >T
Japanese
[32]
c.20131C>T; c.23161A >T
Japanese
[32]
c.8899A>C; c.23908_23911del
Italian
[43]
c.1896+2T>C
Italian
[43]
c.17737-2A>T; c.21423delA
Unknown
[19]
c.24559C>T; c.19429-381_19429-379delinsA
Italian
[43]
c.7310G>A
Italian
[43]
c.22936C>T
Unknown
[19]
c.21840+313_22266 + 5del
Italian
[43]
c.7309C>T
Italian
[43]
c.11086A>C; c.17779_17780delTA
Unknown
[40]
c.10872+1G>T; c.21622A>C
Unknown
[44]
Deletion of exons 14–81 and one-copy loss of exons 82–89
Unknown
[45,46]
c.1152+1G>T; c.11318_11319del
Unknown
[47]
c.20131C>T; c.22924delT
Japanese
[31]
c.21522+3A>G; c.12148G>T
Chinese
[48]
c.23722A>T; c.21522+3A>G
Chinese
Current
Table 2: Summary of NM with NEB mutations: present study and review of literature.
Autosomal recessive mutations in NEB encoding nebulin were reported to cause NM in 1999 [25]. Romero et al. first reported a case with core-rod myopathy caused by NEB gene mutation that showed generalized hypotonia and required immediate intubation and resuscitation at birth [26]. However, clinical phenotypes of NM caused by NEB mutations are variable [27-29]. Some patients have shown mild symptoms and began walking in the normal milestone range or up to 3 years of age [27-29]. However, Rocha et al found a homozygous mutation in NEB intron 122 causing foetal nemaline myopathy with arthrogryposis during early gestation [30]. Previous study reported a NEB-related adult NM patient presenting slowly progressive distal myopathy with respiratory and heart failure. She had a known missense variant of c.20131C > T [31,32], and a novel variant of c.674C>T in NEB [33]. In the present study, we report a childhood-onset NM patient, with suspected positive family genetic history. Whole-exome sequencing and subsequent Sanger sequence analysis for the patient revealed a pathogenic compound heterozygous variant in NEB gene c.21522+3 (IVS 144) A>G and c.23722 (exon 165) A>T. Yin et al reported that splicing change c.21522+3A>G is hotspot mutation in Chinese NM patients [34]. After searching all the literatures, we found that mutation of c.23722 (exon 165) A>T has not been reported yet.
Conclusion
We reported one individual with NM carried new NEB mutation. The results of our study help to expand the mutation spectrum of NEB and enrich the clinical knowledge of this disorder. We suggest that NEB be included in a carrier screening panel in Chinese patients with congenital myopathies.
Author Statements
Acknowledgements
The funding for our study was provided by the National Natural Science Foundation of China (81601041 to J.X, 82171277 to W.J), New Century Talents of Tianjin Medical University General Hospital, Tianjin Municipal Health Commission Science and Technology Project (TJWJ2023QN011 to J.X), Medical Foundation of Jieping (320.6750.19089-56 to C.Y.W).
References
- Sewry CA, Laitila JM, Wallgren-Pettersson C. Nemaline myopathies: a current view. J Muscle Res Cell Motil. 2019; 40: 111-26.
- North KN, Laing NG, Wallgren-Pettersson C. Nemaline myopathy: current concepts. The ENMC international consortium and nemaline myopathy. J Med Genet. 1997; 34: 705-13.
- Pascal A, Caffarri S, Croce R, Sandonà D, Bassi R, Robert B. A structural investigation of the central chlorophyll a binding sites in the minor photosystem II antenna protein, Lhcb4. Biochemistry. 2002; 41: 2305-10.
- Wallgren-Pettersson C, Pelin K, Nowak KJ, Muntoni F, Romero NB, Goebel HH, et al. Genotype-phenotype correlations in nemaline myopathy caused by mutations in the genes for nebulin and skeletal muscle alpha-actin. Neuromuscul Disord. 2004; 14: 461-70.
- Nowak KJ, Wattanasirichaigoon D, Goebel HH, Wilce M, Pelin K, Donner K, et al. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet. 1999; 23: 208-12.
- Agrawal PB, Strickland CD, Midgett C, Morales A, Newburger DE, Poulos MA, et al. Heterogeneity of nemaline myopathy cases with skeletal muscle alpha-actin gene mutations. Ann Neurol. 2004; 56: 86-96.
- Donner K, Ollikainen M, Ridanpàà M, Christen HJ, Goebel HH, de Visser M, et al. Mutations in the beta-tropomyosin (TPM2) gene-- a rare cause of nemaline myopathy. Neuromuscul Disord. 2002; 12: 151-8.
- Eagle LR, Yin X, Brothman AR, Williams BJ, Atkin NB, Prochownik EV. Mutation of the MXI1 gene in prostate cancer. Nat Genet. 1995; 9: 249-55.
- Wattanasirichaigoon D, Swoboda KJ, Takada F, Tong HQ, Lip V, Iannaccone ST, et al. Mutations of the slow muscle alpha-tropomyosin gene, TPM3, are a rare cause of nemaline myopathy. Neurology. 2002; 59: 613-7.
- Agrawal PB, Greenleaf RS, Tomczak KK, Lehtokari VL, Wallgren- Pettersson C, Wallefeld W, et al. Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am J Hum Genet. 2007; 80: 162-7.
- Ravenscroft G, Miyatake S, Lehtokari VL, Todd EJ, Vornanen P, Yau KS, et al. Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am J Hum Genet. 2013; 93: 6-18.
- Gupta VA, Ravenscroft G, Shaheen R, Todd EJ, Swanson LC, Shiina M, et al. Identification of KLHL41 mutations implicates BTB-kelch-mediated ubiquitination as an alternate pathway to myofibrillar disruption in nemaline myopathy. Am J Hum Genet. 2013; 93: 1108-17.
- Yuen M, Sandaradura SA, Dowling JJ, Kostyukova AS, Moroz N, Quinlan KG, et al. Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest. 2014; 124: 4693-708.
- Miyatake S, Mitsuhashi S, Hayashi YK, Purevjav E, Nishikawa A, Koshimizu E, et al. Biallelic mutations in MYPN, encoding myopalladin, are associated with childhood-onset, slowly progressive nemaline myopathy. Am J Hum Genet. 2017; 100: 169-78.
- Johnston JJ, Kelley RI, Crawford TO, Morton DH, Agarwala R, Koch T, et al. A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am J Hum Genet. 2000; 67: 814-21.
- Abdulhaq UN, Daana M, Dor T, Fellig Y, Eylon S, Schuelke M, et al. Nemaline body myopathy caused by a novel mutation in troponin T1 (TNNT1). Muscle Nerve. 2016; 53: 564-9.
- Sandaradura SA, Bournazos A, Mallawaarachchi A, Cummings BB, Waddell LB, Jones KJ, et al. Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum Mutat. 2018; 39: 383-8.
- Feingold-Zadok M, Chitayat D, Chong K, Injeyan M, Shannon P, Chapmann D, et al. Mutations in the NEB gene cause fetal akinesia/arthrogryposis multiplex congenita. Prenat Diagn. 2017; 37: 144-50.
- Lehtokari VL, Kiiski K, Sandaradura SA, Laporte J, Repo P, Frey JA, et al. Mutation update: the spectra of nebulin variants and associated myopathies. Hum Mutat. 2014; 35: 1418-26.
- Malfatti E, Romero NB. Nemaline myopathies: state of the art. Rev Neurol (Paris). 2016; 172: 614-9.
- Wang K, Knipfer M, Huang QQ, van Heerden A, Hsu LC, Gutierrez G, et al. Human skeletal muscle nebulin sequence encodes a blueprint for thin filament architecture. Sequence motifs and affinity profiles of tandem repeats and terminal SH3. J Biol Chem. 1996; 271: 4304-14.
- Lehtokari VL, Pelin K, Sandbacka M, Ranta S, Donner K, Muntoni F, et al. Identification of 45 novel mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Hum Mutat. 2006; 27: 946-56.
- Wang Q, Hu Z, Chang X, Yu M, Xie Z, Lv H, et al. Mutational and clinical spectrum in a cohort of Chinese patients with hereditary nemaline myopathy. Clin Genet. 2020; 97: 878-89.
- Gurgel-Giannetti J, Souza LS, Yamamoto GL, Belisario M, Lazar M, Campos W, et al. Nemaline myopathy in Brazilian patients: molecular and clinical characterization. Int J Mol Sci. 2022; 23: 11995.
- Pelin K, Hilpelà P, Donner K, Sewry C, Akkari PA, Wilton SD, et al. Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci USA. 1999; 96: 2305-10.
- Romero NB, Lehtokari VL, Quijano-Roy S, Monnier N, Claeys KG, Carlier RY, et al. Core-rod myopathy caused by mutations in the nebulin gene. Neurology. 2009; 73: 1159-61.
- Malfatti E, Monges S, Lehtokari VL, Schaeffer U, Abath Neto O, Kiiski K, et al. Bilateral foot-drop as predominant symptom in nebulin (NEB) gene related ”core-rod” congenital myopathy. Eur J Med Genet. 2015; 58: 556-61.
- Scoto M, Cullup T, Cirak S, Yau S, Manzur AY, Feng L, et al. Nebulin (NEB) mutations in a childhood onset distal myopathy with rods and cores uncovered by next generation sequencing. Eur J Hum Genet. 2013; 21: 1249-52.
- Wunderlich G, Brunn A, Daimagüler HS, Bozoglu T, Fink GR, Lehmann HC, et al. Long term history of a congenital core-rod myopathy with compound heterozygous mutations in the nebulin gene. Acta Myol. 2018; 37: 121-7.
- Rocha ML, Dittmayer C, Uruha A, Korinth D, Chaoui R, Schlembach D, et al. A novel mutation in NEB causing foetal nemaline myopathy with arthrogryposis during early gestation. Neuromuscul Disord. 2021; 31: 239-45.
- Tsunoda K, Yamashita T, Motokura E, Takahashi Y, Sato K, Takemoto M, et al. A patient with slowly progressive adult-onset nemaline myopathy and novel compound heterozygous mutations in the nebulin gene. J Neurol Sci. 2017; 373: 254-7.
- Mizuno Y, Mori-Yoshimura M, Oya Y, Nishikawa A, Nishino I, Takahashi Y. [Two cases of nemaline myopathy presenting with hypertrophy of distal limbs with prominent asymmetry]. Rinsho Shinkeigaku. 2017; 57: 691-7.
- Ohara M, Saito Y, Watanabe M, Mizutani S, Kobayashi M, Iida A, et al. An adult nemaline myopathy patient with respiratory and heart failure harboring a novel NEB variant. eNeurologicalSci. 2020; 21: 100268.
- Yin X, Pu C, Wang Z, Li K, Wang H. Clinico-pathological features and mutational spectrum of 16 nemaline myopathy patients from a Chinese neuromuscular center. Acta Neurol Belg. 2022; 122: 631-9.
- Mubaraki AA. Nemaline myopathy: A case report. Case Rep Neurol. 2021; 13: 499-503.
- Ma Y, Zhang H, Li X, Yang X, Li Y, Guan J, et al. An integration-free iPSC line (SDQLCHi017-A) derived from a patient with nemaline myopathy-2 disease carrying compound heterozygote mutations in NEB gene. Stem Cell Res. 2020; 43: 101729.
- Laflamme N, Lace B, Thonta Setty S, Rioux N, Labrie Y, Droit A, et al. A homozygous deep intronic mutation alters the splicing of nebulin gene in a patient with nemaline myopathy. Front Neurol. 2021; 12: 660113.
- Zhang Y, Yan H, Liu J, Yan H, Ma Y, Wei C, et al. Clinical and genetic features of infancy-onset congenital myopathies from a Chinese paediatric centre. BMC Pediatr. 2022; 22: 65.
- Zhang YL, Zhen L, Xu LL, Li DZ. Fetal akinesia: the need for clinical vigilance in first trimester with decreased fetal movements. Taiwan J Obstet Gynecol. 2021; 60: 559-62.
- Moreau-Le Lan S, Aller E, Calabria I, Gonzalez-Tarancon L, Cardona-Gay C, Martinez-Matilla M, et al. New mutations found by Next-Generation Sequencing screening of Spanish patients with nemaline myopathy. PLOS ONE. 2018; 13: e0207296.
- Nampoothiri RV, Jain A, Malhotra P. Reversible proptosis due to a hematological cause. Hematol Transfus Cell Ther. 2018; 40: 196-7.
- Huang K, Luo YE, Li QX, Duan HQ, Bi FF, Yang H et al. Clinical, pathological and genetic studies of two cases of childhood-onset nemaline myopathy. Zhongguo Dang Dai Er Ke Za Zhi. 2018; 20: 804-8.
- Piga D, Magri F, Ronchi D, Corti S, Cassandrini D, Mercuri E, et al. New mutations in NEB gene discovered by targeted next-generation sequencing in nemaline myopathy Italian patients. J Mol Neurosci. 2016; 59: 351-9.
- Güttsches AK, Dekomien G, Claeys KG, von der Hagen M, Huebner A, Kley RA, et al. Two novel nebulin variants in an adult patient with congenital nemaline myopathy. Neuromuscul Disord. 2015; 25: 392-6.
- Lawlor MW, Ottenheijm CA, Lehtokari VL, Cho K, Pelin K, Wallgren-Pettersson C, et al. Novel mutations in NEB cause abnormal nebulin expression and markedly impaired muscle force generation in severe nemaline myopathy. Skelet Muscle. 2011; 1: 23.
- Kiiski KJ, Lehtokari VL, Vihola AK, Laitila JM, Huovinen S, Sagath LJ, et al. Dominantly inherited distal nemaline/cap myopathy caused by a large deletion in the nebulin gene. Neuromuscul Disord. 2019; 29: 97-107.
- Piteau SJ, Rossiter JP, Smith RG, MacKenzie JJ. Congenital myopathy with cap-like structures and nemaline rods: case report and literature review. Pediatr Neurol. 2014; 51: 192-7.
- Wen Q, Chang X, Guo J. A childhood-onset nemaline myopathy caused by novel heterozygote variants in the nebulin gene with literature review. Acta Neurol Belg. 2020; 120: 1351-60.