
Case Report
Austin J Clin Case Rep. 2023; 10(8): 1308.
Eosinophilic Granulomatosis with Polyangiitis Revealed by Acute Cholecystitis: A Rare Manifestation
Yassine Ennaboulsi¹*; Salah Eddine E¹ Khader1; Mehdi El Aissate¹; Amine Kessab²; Mohammed Karim Moudden¹; Ali Zinebi¹
¹Department of Internal Medicine, Moulay Ismail Military Hospital, Boulevard El Hanssali, Meknes, Morocco
²Department of Pathology, Moulay Ismail Military Hospital, boulevard El Hanssali, Meknes, Morocco
*Corresponding author: Yassine Ennaboulsi Department of Internal Medicine, Moulay Ismail Military Hospital, Boulevard El Hanssali, 50000, Meknes, Morocco Te: +212 6 06 59 12 87 Email: ennaboulsiyass@gmail.com
Received: October 18, 2023 Accepted: November 11, 2023 Published: November 18, 2023
Abstract
Acute cholecystitis is a rare disease of eosinophilic granulomatosis with polyangiitis, which is a systemic necrotizing vasculitis affecting small- to medium-sized vessels, associated with blood and tissue eosinophilia and the presence of extravascular granulomas. We report the case of a 53-year-old female patient who presented with acute acalculous cholecystitis who underwent surgery. The diagnosis of eosinophilic granulomatosis with polyangiitis was based on histology of the gallbladder, antineutrophil cytoplasmic antibody positivity and the presence of polyneuropathy. Corticosteroids allowed a favorable clinical outcome with no relapses during follow-up.
Keywords: Eosinophilic granulomatosis with polyangiitis; Acute cholecystitis; Antineutrophil cytoplasmic antibody; Corticosteroid; Pathology
Abbreviations: EGPA: Eosinophilic granulomatosis with polyangiitis, ANCA: Anti-neutrophil cytoplasmic antibodies
Introduction
Eosinophilic Granulomatosis with Polyangiitis (EGPA), formerly known as Churg-Strauss syndrome, is a systemic necrotizing vasculitis affecting small- to medium-sized vessels, associated with blood and tissue eosinophilia and the presence of extravascular granulomas [1]. It is a rare condition with an annual incidence of 0.9 to 2.4 cases per million population [2].
Anti-Neutrophil Cytoplasmic Antibodies (ANCA) are positive in only around 30% of cases, which leads us to believe that this is a heterogeneous disease with two phenotypes depending on the presence or absence of ANCA.
It can affect the peripheral nerves, skin, lungs, kidneys, cardiovascular system and gastrointestinal tract, but acute cholecystitis is a rare disease of EGPA [3], which makes diagnosis difficult.
We report here a rare case of EGPA manifested by acute acalculous cholecystitis.
Case Report
A 53-year-old patient with no significant history of illness had been hospitalized for recurrent fever and occasional epigastric pain for two weeks.
Abdominal examination revealed tenderness over the right hypochondrium and Murphy's sign was positive. Laboratory investigations showed the following results: white blood cell count 17230/mm3 including 15080/mm³ of neutrophils and 240/mm3 of eosinophils, C-Reactive Protein (CRP) of 230.58mg/L, aspartate aminotransferase of 22IU/ L, alanine aminotransferase of 10IU/L, total bilirubin of 4.11mg/L, Gamma-glutamyl transferase of 72IU/L and alkaline phosphatase of 235IU/L.
Abdominal ultrasonography and abdominal computed tomography revealed a parietal thickening of the gallbladder without visible lithiasis (Figure 1A,1B).

Figure 1A: Abdominal ultrasonography revealed parietal thickening of the gallbladder without visible lithiasis.

Figure 1B: Abdominal computed tomography revealed thickened gallbladder with oedema but no evidence of lithiasis.
The diagnosis of acute acalculous cholecystitis was made and the patient was given antibiotics, but there was no clinical improvement in the first 24 hours, so a cholecystectomy was performed.
A week after surgery, she presented with numbness in both upper limbs and was referred to our department. The neurological examination found a predominant sensitive polyneuropathy in the upper limbs, the abdominal examination was normal except for the visible cholecystectomy scar. The rest of the clinical examination, in particular the pleuropulmonary, cardiovascular examinations was without particularity. Laboratory investigations showed that white blood cell count 6660/mm3 including 70/mm3 of eosinophils, C-Reactive Protein (CRP) of 130,80mg/L, aspartate aminotransferase of 22IU/L, alanine aminotransferase of 7IU/L, Gamma-glutamyl transferase of 52IU/L and alkaline phosphatase of 183IU/L.
Gallbladder pathology returned showing an inflammatory infiltrate rich in eosinophils at the serous (Figure 2A,2B,2C).
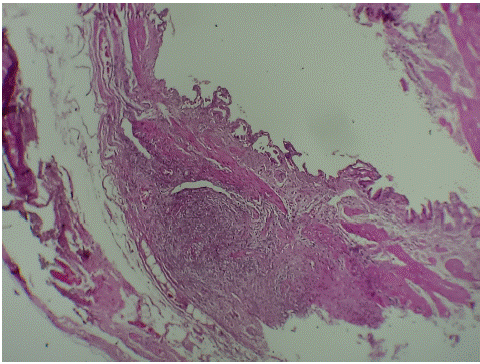
Figure 2A: Histological image showing a vesicular wall with an inflammatory infiltrate in the subserosa. magnification×10.

Figure 2B: Histological image showing polymorphism of inflammatory cells with vascular congestion, magnification×20.
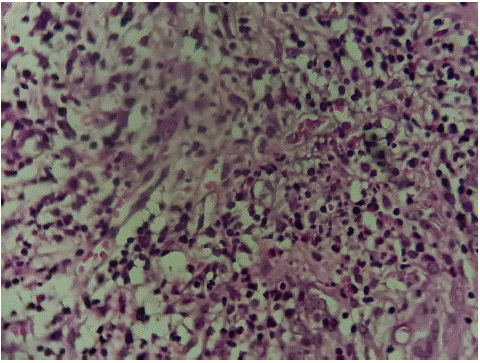
Figure 2C: Histological image showing the presence of moderate amounts of eosinophils within the inflammatory infiltrate. Magnification×40.
Electrodiagnostic study showed sensory-motor axonal neuropathy of the four limbs. The immunological evaluation found the myeroperoxidase antineutrophil cytoplasmic antibody (MPO-ANCA) was present, Chest x-ray showed no pulmonary infiltrates.
The diagnosis of eosinophilic granulomatosis with polyangiitis was based on the presence of polyneuropathy, eosinophilic infiltration of the gallbladder and the presence of ANCA.
Treatment was started with prednisone at an initial dose 60 mg daily, and the response to treatment was good, with rapid improvement in clinical symptoms. No relapses were observed during follow-up.
Discussion
EGPA is a rare granulomatous necrotizing vasculitis, characterized by diffuse involvement of small -to medium-sized vessels with eosinophilic infiltration, first described by Churg and Strauss in 1951 [4].
It can manifest by various symptoms related to peripheral neuropathy, myocarditis, glomerulonephritis, purpuric lesions of the skin. Cardiac manifestations tend to be the leading cause of death, accounting for up to 48%.
Gastrointestinal involvement occurs in 32.6% of cases [5]. It may precede or occur at the same time as the vasculitis phase, often due to eosinophilic infiltration of the gastrointestinal mucosa. Sometimes there may be ischemic lesions due to vasculitis, especially in the small intestine, which may lead to digestive perforation and bleeding [6].
Acute cholecystitis is a rare disease of EGPA [7], only 15 cases of EGPA patients with acute cholecystitis were reported in the literature. In a large cohort of patients with GEPA, by Guillevin et al, only one in 96 had developed cholecystitis in the 32 years of follow-up [8].
The American College of Rheumatology (ACR) has proposed criteria for the diagnosis of EGPA (asthma, transient pulmonary infiltrates, eosinophilia >10% on differential, Mono- or polyneuropathy, Nasal sinusitis and Biopsy showing vasculitis lesions with eosinophilic infiltrate). The presence of four of the six elements has a sensitivity of 85% and a specificity of 99.7% for the diagnosis of EGPA [9].
The diagnosis is essentially clinical, the presence of ANCA contributes to the diagnosis but they can be absent so it is the pathology when the biopsy is performed that represents a strong argument for the diagnosis of EGPA. Typical pathological changes are eosinophilic infiltration, granulomatous inflammation and necrotizing vasculitis. The main pathological features of the gallbladder are eosinophilic infiltration and necrotizing vasculitis of small-to medium-sized vessels [10,11]. In our case it was the neurological damage, the pathology and the presence of the ANCA that made it possible to establish the diagnosis even in the absence of the other criteria including asthma, allergic history and blood eosinophilia.
Treatment of non-severe EGPA is based on corticosteroid therapy, which is generally effective; however, in the case of a poor response or relapse, additional immunosuppressive treatment is required [12]. In severe forms with major organ involvement, a combination of corticosteroid therapy and an immunosuppressive therapy, mainly cyclophosphamide, is recommended [13]. Corticosteroid alone was effective in our patient, without notion of relapse.
Recently, it has been shown that mepolizumab, an anti-IL 5 antibody, is potentially effective as an induction therapy in patients with severe EGPA and may be useful in reducing doses of corticosteroids [14].
Conclusion
We report here a rare case of EGPA manifested by acute cholecystitis and whose diagnosis was made by pathology even in the absence of typical manifestations, surgeons and clinicians should not overlook the gastrointestinal and neurological involvement of EGPA in order to make the diagnosis quickly and start treatment early to ensure a good prognosis.
Author Statements
Conflicts of Interest
There is no Conflict of Interest inside this manuscript.
References
- Fagni F, Bello F, Emmi G. Eosinophilic granulomatosis with polyangiitis: dissecting the Pathophysiology. Front Med (Lausanne). 2021; 8: 627776.
- Furuta S, Iwamoto T, Nakajima H. Update on eosinophilic granulomatosis with polyangiitis. Allergol Int. 2019; 68: 430-6.
- Ohnuki Y, Moriya Y, Yutani S, Mizuma A, Nakayama T, Ohnuki Y, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) complicated by perforation of the small intestine and cholecystitis. Intern Med. 2018; 57: 737-40.
- Churg J, Strauss L. Allergic angiitis and periarteritis nodosa. Am J Pathol. 1951; 27: 277-301.
- He JN, Tian Z, Yao X, Li HY, Yu Y, Liu Y, et al. Multiple perforations and fistula formation following corticosteroid administration: a case report. World J Clin Cases. 2017; 5: 67-72.
- Pagnoux C, Mahr A, Cohen P, Guillevin L. Presentation and outcome of gastrointestinal involvement in systemic necrotizing vasculitides: analysis of 62 patients with polyarteritis nodosa, microscopic polyangiitis, Wegener granulomatosis, Churg-Strauss syndrome, or rheumatoid arthritis-associated vasculitis. Med (Baltim). 2005; 84: 115-28.
- Ye L, Lu X, Xue J. Eosinophilic granulomatosis with polyangiitis complicated by cholecystitis: A case report and review of the literature. Clin Rheumatol. 2016; 35: 259-63.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Med (Baltim). 1999; 78: 26-37.
- Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 1990; 33: 1094-100.
- Lenders G, Goethals M, Verstreken S, Dierckx R, Vanderheyden M. Acalculous cholecystitis and tamponade: an unusual combination? Acta Cardiol. 2011; 66: 383-5.
- Sironen RK, Seppä A, Kosma VM, Kuopio T. Churg-Strauss syndrome manifested by appendicitis, cholecystitis and superficial micronodular liver lesions–an unusual clinicopathological presentation. J Clin Pathol. 2010; 63: 848-50.
- Doubelt I, Pulenzas N, Carette S, Pagnoux C, Canadian Vasculitis Network (CanVasc). Efficacy of conventional immunosuppressants in relapsing or refractory eosinophilic granulomatosis with polyangiitis: evidence from a Canadian single-centre cohort. Clin Exp Rheumatol. 2020; 124: 171-5.
- Pagnoux C, Groh M. Optimal therapy and prospects for new medicines in eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). Expert Rev Clin Immunol. 2016; 12: 1059-67.
- Hattori K, Teramachi Y, Kobayashi Y, Ito T, Morinaga T, Tamai H, et al. A case of effective mepolizumab induction therapy for severe eosinophilic granulomatosis with polyangiitis diagnosed by eosinophilic cholecystitis and interstitial nephritis. Case Rep Rheumatol. 2021; 2021: 1-5.