
Case Report
Austin J Clin Case Rep. 2024; 11(1): 1314.
Thoracic Spinal Cord Primary Mesenchymal Chondrosarcoma. Case Report
Martha Lilia Tena-Suck1; Nora Kerik2; Oriana Carolina García-Diaz3; Ivan Eudaldo Diaz Meneses2; Juan Salvador Rosales García2; Samuel Ismael Juárez Cruz1; Nicasio Arriaga4
1Department of Neuropathology, National Institute of Neurology and Neurosurgery, Mexico
2Molecular Imaging Unit PET-CT, National Institute of Neurology and Neurosurgery, Mexico
3Hospital General Secretaria de Salud 450 Durango, Durando
4Department of Neuropathology, Service of Spinal Neurosurgery, National Institute of Neurology and Neurosurgery, Mexico
*Corresponding author: Martha Lilia Tena-Suck, MD Department of Neuropathology, National Institute of Neurology and Neurosurgery, México City, Av. Insurgentes sur no 3877, Colonia la joya, Delegación Tlalpan, Cuidad de México. Email: mltenasuck@gmail.com
Received: February 02, 2024 Accepted: March 13, 2024 Published: March 20, 2024
Abstract
Background: Mesenchymal chondrosarcoma is a rare, high-grade malignancy of bone or soft tissue with a unique biphasic histology and worse prognosis. It rarely occurs in the spinal region.
Clinical Case: We present the case of a 21-year-old woman who became pregnant and began with feeling of heavy legs and decreased strength. who progressively over a week was unable to walk, was scheduled for cesarean section and the patient showed complete loss of sensation, and urinary incontinence. Spinal TAC and PET showed a destructive tumor in T11-L1. She underwent laminectomy and a biphasic lesion was identified, that forming by a component of small round, blue cells, hyper vascularized with nests of cartilage interspersed between these cells. By immunohistochemistry it was positive for CD99, vimentin, focally for-s1-00 and IDH1 in the round cells and in the periphery of the cartilaginous areas. Weak synaptophysin immunoexpression and higher Ki67 and p53 li. Among the differential diagnoses were Edwing sarcoma, medulloblastoma, and small cell osteosarcoma.
Discussion: The origin of this tumor and especially the biphasic component are discussed. That could correspond to a tumor of immature cells of the stem cell type with different chondromatous maturation processes.
Keywords: Mesenchymal chondrosarcoma; Spinal chondrosarcoma; Spinal tumors; Immunohistochemistry; Positron Emission Tomography (PET).
Abbreviations: MCS: Mesenchymal Chondrosarcoma; WHO: World Health Organization; STT: Soft Tissue Tumors; CBTs: Bone Classifies Chondrogenic Bone Tumors; ACTs: Atypical Cartilaginous Tumors; CS: Chondrosarcoma; IDH1 and IDH2: Isocitrate Dehydrogenase 1 and 2 Mutations; ECM: Eosinophilic Chondroid Matrix; EMCS: Extraskeletal Mesenchymal Chondrosarcoma; EMA: Epithelial Membrane Antigen; PET: Positron Emission Tomography
Background
Chondrosarcoma (CS) is a malignant cartilaginous tumor that can be histologically categorized into 3 types: mesenchymal, classic, and myxoid [1]. In conclusion, the 2020 World Health Organization (WHO) classification classifies chondrosarcomas into eight subtypes: central conventional (grade 1 vs. 2–3), secondary peripheral (grade 1 vs. 2–3), periosteal, dedifferentiated, mesenchymal, and clear cell [2]. Intracranial MSCs can be characterized into 3 grades: Grade 1 (well differentiated), Grade 2 (moderately differentiated), and Grade 3 (poorly differentiated). Several subtypes exist that fluctuate in MCS [1-4], it is a rare soft tissue tumor arising from soft tissues, further most commonly originating in the bone, in extraskeletal sites has also been presented [2,3,5].
MCS is a well-defined tumor entity first described in 1959 by Lichtenstein and Bernstein [6] and Dahlin and Henderson [7]; in 1962 described 9 cases from the files of the Mayo Clinic.
MCS occurring principally of the lower extremities, meninges, and orbits, has a slight predominance in females with a general poor prognosis, and affects all ages, with greater frequency in the second decade of life [3], and has a variable clinical course with frequent recurrences and occasional distant osseous and visceral metastatic spread [1]. MCS is a rare malignant variant of chondrosarcoma whose incidence accounts for 0.2–0.7% of all malignant bone tumors or 3–10% of CS [3,4]. They are usually bone tumors but can be detected in the extra-skeletal sites. Extra-skeletal MCSs most often involve the brain and meninges, occasionally the intraspinal region [1]. Young adults are more susceptible to developing Extraskeletal Mesenchymal Chondrosarcoma (EMCS), MCS is generally grossly lobulated, firm, with ossified or cartilaginous elements, and often hypervascularity. MCS is morphologically categorized by a biphasic pattern of small round cells and islands of well-differentiated hyaline cartilage and composed of an admixture of undifferentiated mesenchymal cell cartilage [2-4].
Histopathological shows that the tumor is composed of spindle and round cells with a high nucleocytoplasmic ratio accompanied by scattered eosinophilic chondroid matrix or nest of cartilage [1-4].
MCS molecular features are similar to is a totally different pathological than the conventional Chondrosarcoma, this entity exhibiting complex cytogenetic alterations. HEY1-NCOA2 (8;8) (q21; q13) fusion is most described in MCS [4]. Other genes implicated have been IRF2BP2 gene and the transcription factor CDX1 gene [5].
The aim of this case reports a rare case of Mesenchymal chondrosarcoma origin in thoracic spinal cord in a 38 years old-man with weak IDH1 immunoexpression.
Clinical Case
We present the case of a 21-year-old woman who became pregnant and began with heavy legs and decreased strength, which progressively, over a week, was unable to walk. She was scheduled for cesarean section and the patient showed with hypoesthesia in the legs, complete loss of sensation and complete inability to walk and without strength with urinary incontinence. Acute myelitis was diagnosed, to the admission to our institution. The cerebrospinal fluid showed pleocytosis, and an MRI and PET analysis showed a lesion at T11-L1 (Figure 1). Positron Emission Tomography (PET) with Fluoro desoxiglucose 18FDG (10 Mci) whole body and brain scan were performed a (Figure 4) at 60 minutes post radiotracer administration, the scan was obtained on the ™ Biograph 64 mCT (Siemens Healthcare Molecular Imaging) hybrid device [12]. The images were reconstructed using the iterative method UltraHD PET (Siemens ™), obtaining a clinical spatial resolution of 4 mm. The images were visualized in a multimodal medical-grade workstation equipped with SYNGO software (Siemens ™) and qualitatively analyzed by two nuclear medicine physicians. Which showed: Ovoid, hyperdense lesion, with heterogeneous density at the expense of some calcifications, dependent on the T12 nerve root, protruding through the foramen at this level with approximate measurements of 20x15x20 mm which contacts the dura mater, reaching the adjacent paravertebral region, with an increase in SUVmax metabolism of 7.2. There were no other relevant morphological findings in the oncological clinical context.
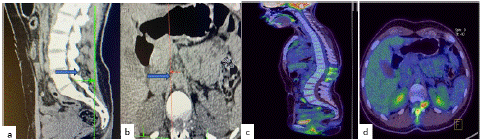
Figure 1: (a) Sagittal TAC Imaging showed a lesion in T11-12, (b) Axial TAC Imaging showed the spinal lesion. (c, d) PET-CT FDG, sagittal, and axial Which showed: Ovoid, hyperdense lesion, with heterogeneous density at the expense of some calcifications, dependent on the T12 nerve root, (d) protruding through the foramen at this level with approximate measurements of 20x15x20 mm which contacts the dura mater, reaching the adjacent paravertebral region, with an increase in SUVmax metabolism of 7.2.
Patients was undergoing laminectomy. Tumor corresponding an ovoid lesion was received that measured 25x20mm. It was reddish with a soft surface; the cut showed numerous dilated blood vessels with a hemorrhagic appearance.
Histologically, a neoplasm formed by small, round, blue, and hyperchromatic cells with a densely vascularized and hemorrhagic background is observed (Figure 2a), interspersed with nests of mature cartilage or Eosinophilic Chondroid Matrix (ECM) (Figure 2b and 2c), hyalinized stroma (Figure 2d), also foci of necrosis (Figure 2e), dystrophic classifications (Figure 2f) and bone metaplasia were observed (Figure 2g). Cellular atypia was observed in rounds cells as well as in eosinophilic chondroid matrix (Figure 2f). Cellular atypia and mitosis figures were observed in round neoplastic cells (Figure 2g) and in the cartilage cells (Figure 2h). PAS staining, the ECM seen in pink were evident (Fig 2i), and reticular fibers stain showed a fibrous stroma (Figure 2j)
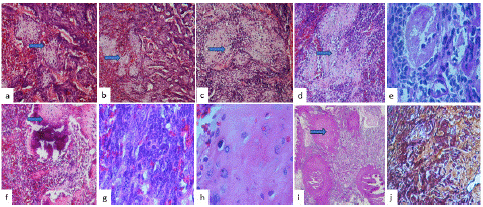
Figure 2: Histological features photographs (a), showed a tumor formed by small, round, blue, and hyperchromatic cells with a densely vascularized and hemorrhagic background is observed interspersed with nests of mature cartilage in (b) and c), hyalinized stroma around the nest of cartilage in (d)(H&Ex200), necrosis (e), dystrophic classifications and bone metaplasia were observed (f). Cellular atypia was observed in round cells (g) and in eosinophilic chondroid matrix (h)(H&Ex400). PAS staining, the eosinophilic chondroid matrix is evident in pink stain (i) (PAS x200), and the reticular fibers stain showed a fibrous stroma (RS x400).
Immunohistochemical staining was performed, with the neoplastic cells being slightly cytoplasmically positive for s-100 (Figure 3a), synaptophysin (Figure 3b), osteoponin, osteoconnectin (Figure 3c), vimentin (Figure 3d), and CD99 and ureweak immunoreaction to IDH1 (Figure 3f. The mib-1(ki67) index was 50% and was intensely positive for p53 (Figure 3g), CD34 were strongly positive in the blood vessel wall and tumor (Figure 3h). Tumor cells were negative for EMA, GFAP, CK8 EMA, chromogranin, myogenin, and myoD1, CD45, desmin, Nestin, CD45 and ENE, chromogranin, CK8, INI1 and Stat6 were negative, based on the histological appearance and immunohistochemistry results, it was diagnosed as spinal mesenchymal chondrosarcoma.
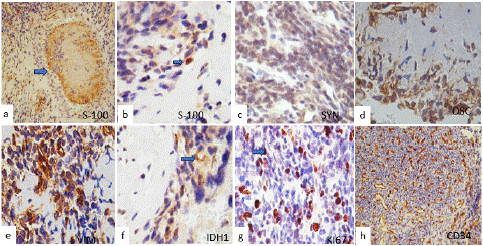
Figure 3: Immunohistochemical stain. (a) and (b) Showed the neoplastic cells cytoplasmically positive in a few cells for s-100 and in the peripheral region of the eosinophilic chondroid matrix. Tumor cells were also positive for synaptophysin in (c), osteonectin (d), vimentin (e), and IDH1 (f). Ki67 was intensely positive immunoreaction in (g), and CD34 was strongly positive in the blood vessel wall and tumor in (h) (original magnifications x400). The blue arrows showed positive immunoexpression in the peripheral region of the eosinophilic chondroid matrix.
Discussion
MCS is a rare bone tumor, especially intramodular location. Since in 2019 they had been reported in the medical literature clinical information for 18 patients with primary intradural MCS [8].
MRI remains the preferred imaging modality for intraspinal tumors, but there is no pathognomonic description for extra-osseous MCS. However, extra-osseous MCS typically present isointense signals with respect to the normal spinal cord on T1- weighted images while T2-weighted images show a high intensity or isointensity [9].
The MRI imaging of chondrosarcomas vary meaningfully depending upon the histologic grade. The spectrum of findings starts with lysis, which is difficult to discriminate between enchondromas and chondrosarcomas [3]. High-grade tumors are demonstrated radiographically with moth-eaten destruction and intermittent periosteal reaction. Higher differentiation is related to the presence of a “rings and arcs” pattern of calcification into the tumor matrix [4]. The differential diagnosis depending on the presence of calcifications and nonetheless, is believed to be not significantly related to the histologic findings and prognosis [9]. Some variant subtypes of CS are recognized, which are extremely rare, especially when originating in the spine [9].
The literature has reported the ability of 18FFDG PET-CT avidity to make a distinction (diagnosis, therapeutic strategy, invasive procedures, and percutaneous biopsy) between benign cartilaginous lesions and high chondrosarcoma [10]. The overall 18FFDG PET/CT has a higher accuracy to differentiate between them. Some authors proposed a cutoff value of SUVmax 2.6 and 2.0. (In a meta-analysis was reported the relatively accurate diagnostic efficacy of 18F-FDG PET (sensitivity = 0.84; specificity = 0.82) and PET/CT (sensitivity = 0.94; specificity = 0.89) for the diagnosis of chondrosarcoma [10]. SUVmax values have been observed to be increased with the tumor grade [10]. In our case, the SUV max of the lesion was 7.2. Intraoperatively and radiologically, MCs can be misdiagnosed as atypical meningioma, malignant meningiomas, hemangiopericytoma, schwannomas, metastasis, gliomas, or oligodendroglioma [1,5].
MCS is a high-grade malignant tumor with a robust propensity for locally recurrent or distant metastasized. The prognosis of MCS is bad and very variable; it varies depending on the location of the tumor and tumor size. They have high morbidity and mortality rates [1,5,7], and about their malignancy and progressiveness, MCs are considered as a distinct entity that is entirely different from the classical chondrosarcomas [7].
Histologically, most MCSs exhibit a biphasic pattern of islands of cartilage and areas of neoplastic, small, round, and blue cell components [5]. The precise histogenesis of intradural chondrosarcomas is still questionable. A likely hypothesis states that chondrosarcomas originate from primitive multipotential mesenchymal cells. Lesions can contain bone or cartilage matrix as an incidental, often metaplastic phenomenon or diagnostic feature. Associating imaging findings with pathology is required to confirm that a tumor-creating bone or cartilage, in detail, invents occurring from soft tissue rather than from the skeleton. Unlike matrices existing in bone tumors where they likely divulge the respective cells of origin (i.e., osteoblastic or chondroblasts precursors), those could be present in soft tissue tumors more often mean a metaplastic phenomenon and reproduce the diversity of differentiation. These tumors can present. These tumor types include the ossifying fibromyxoid tumor, phosphaturic mesenchymal tumor, synovial enchondromatosis, soft tissue chondroma, calcifying aponeurotic fibroma, giant cell tumor of soft tissue, myositis ossificans, and related diseases, mesenchymal chondrosarcoma, and extraskeletal osteosarcoma [11]. MCS has histological features distinguishing biphasic histology comprising the tumor is forming by round blue or spindled cells arranged in small clusters or around blood vessels in a hemangiopericytomatous pattern and the differentiated cartilage [5].
Sheets of primitive mesenchymal cells with scattered islands of well-differentiated hyaline cartilage or eosinophilic chondroid matrix. The cartilaginous foci are usually well circumscribed with a well-defined interface with undifferentiated cells, or can rarely have poorly defined borders that gradually merge with the undifferentiated tumor cells, also, foci of osteoid formation and calcification can be seen [5]. Our case showed small round cells with clear-appearing cells, a dense solid pattern alternating with areas of hemangiopericytomatous or hypervascular appearance with sinusoidal-looking vessels [5]. These small, round, and blue cells showed more undifferentiated or smaller hyperchromatic areas; we observed nests of cartilage that he also knows what their name is eosinophilic chondroid matrix in different stages of differentiation as well as foci of calcification and a discrete material between the vessels that we suggest chondroid material, also nodes of collagenization were observed [5]. By immunohistochemistry, these tumor cells usually express s100 protein, vimentin; CD99, synaptophysin, and Bcl2 [1,2]. In our case, immature small cells were negative for all markers, focally or weak immunoexpression for s-100. While the peripheral areas of the cartilage samples or eosinophilic chondroid matrix were focally positive, immunoreaction for S-100, osteonectin, and osteopontin, and CD34 was positive in the blood vessels. Ki67 and p53 were strong expression. The PAS stain showed strong pink stain in ECM and with the RF stain observed dense fibrillar and collagenized areas. The diagnosis of small round cell tumors always has been extremely difficult, and our current classification systems continue to evolve. The histological differential diagnosis for MCS includes synovial sarcoma, malignant solitary fibrous tumor, Ewing sarcoma, and other small, round blue cell tumors, like as, However, they also may include other tumors such as desmoplastic small round cell tumor, and small cell osteosarcoma, synovial sarcoma, or small cell osteosarcoma particularly in small biopsy specimens [12].
Among the differential diagnoses that we must make is with small, blue and round cell tumors of childhood with: lymphoma (CD45+), Edwing sarcoma (granular intracytoplasmic PAS, EWSR1/FUS and ETS family of transcription factors gene fusion), PNET (synaptophysin (+), medulloblastoma (synaptophysin and ENE+). Although the cells were s-100+ the staining is weak and focal in isolated cells. Staining for osteoponin, osteonectin and vimentin were positive in the cytoplasm of the neoplastic cells and were also found in the cartilage nests in both the central and lateral portions. peripheral of the cartilage nests or ECM and in dystrophic calcifications with bone metaplasia. These tumors are malignant with uncertain clinical behavior, so we observed intensely positive ki67 and p53 indexes [13]. Similar to other chondrosarcomas, the cartilaginous component is usually strongly positive immunoexpression for S100 protein. Occasionally Positive in a cartilaginous component. While, the undifferentiated cells show scant patchy positivity for S100 protein and Sox9 [12]. Furthermore, cytokeratin, Epithelial Membrane Antigen (EMA), and muscle markers are regularly negative immunoexpression in CS. And in MCS [13,14]. Nonetheless, rare published cases showing dispersed and focal tumor cells positive reaction to desmin, myogenin, and myoD1, those results are described as rhabdomyosarcomata’s differentiation [15, 16]. However, IDH1 and IDH2 mutations have not been distinguished in MCS [17]. Usually is positive in CS However, in our case there was a weak expression of IDH1 in some cells. The transcription factor Sox9 has been demonstrated to be a master regulator of the differentiation of mesenchymal cells into chondrocytes [16]. Fanburg-Smith et al. [18] suggested that this type of small-cell tumor results from primitive chondroprogenitor cells from other primitive small-cell malignancies. Therefore, has phenotypic features corresponding to the early condensational phase of cartilaginous differentiation. More significant, Sox9 could be as a useful instrument in the differentiation of small, round and blue cell malignancies [15,16].
Sox9 is a regulator of chondrogenesis, beta-catenin is involved in bone formation, believed to inhibit chondrogenesis in a Sox9-dependent manner, and osteocalcin is an important marker for osteoblastic phenotype [18]. The protein Sox9 can serve as a discriminative marker to differentiate MCS from other small blue round cell tumors. Fanburg-Smith et al. [18] suggest that this component of mesenchymal chondrosarcoma may be a differentiated (benign or metaplastic) element of a malignant metastasizing tumor. This hyaline cartilage is morphologically different from classical chondrosarcoma's cartilage and small-cell osteosarcoma [18].
In another hang, in our case, desmin, myogenin, myoD1, CD45, synaptophysin, chromogranin, CK8, and Stat6 were negative, so we ruled out medulloblastoma, Ewing sarcoma, Ewing-like sarcomas, metastasis, rhabdomyosarcoma, solitary fibrous tumor, lymphoma, etc. [4,5]. Considering them as differential diagnoses due to having small undifferentiated cells. The crucial histopathologic assessment plays a key in guiding appropriate management strategies and enhancing patient outcomes. Conclusion: In the present work, we presented a rare case of mesenchymal sarcoma or wrongly called mesenchymal chondrosarcoma, in a young female. Histologically, it drew attention that this tumor presented a hemangioblastomatous pattern, with discrete osteoid formation, cartilage nests or eosinophilic chondroid matrix in different stages of maturation, or differentiation and fibrotic stroma is also seen.
Author Statements
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Ethical approval to report this case was obtained from mother of the patient.
References
- Fletcher DM, Unni KK, Mertens F. World Health Organization classification of tumours. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press. 2002.
- Nakashima Y, de Pinieux G, Ladanyi M. Mesenchymal condrosarcoma In Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds Pathology and Genetics of Tumours of Soft Tissue and Bone Lyon, France IARC Press. World Health Organization Classification of Tumours. 2013; 5: 271-272.
- Choi JH, Ro JY. The 2020 WHO Classification of Tumors of Bone: An Updated Review. Adv. Anat. Pathol. 2021; 28: 119-138.
- Wang L, Motoi T, Khanin R, Olshen A, Mertens F, Bridge J, et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer. 2012; 51: 127-139.
- Nyquist KB, Panagopoulos I, Thorsen J, Haugom L, Gorunova L, Bjerkehagen B, et al. Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1 fusion gene brought about by translocation t (1;5) (q42; q32) in mesenchymal chondrosarcoma. PLoS One. 2012; 7: e49705.
- Lichtenstein L, Bernstein D. Unusual benign and malignant chondroid tumors of bone: a survey of some mesenchymal cartilage tumors and malignant chondroblastic tumors, including a few multicentric ones, as well as many atypical benign chondroblastomas and chondromyxoid fibromas Cancer 1959; 12: 1142- 1157.
- Dahlin DC, Henderson ED. Mesenchymal chondrosarcoma: further observations on a new entity Cancer 1962; 15: 410-417.
- Chen CW, Chen IH, Hu MH, Lee JC, Huang HY, Hong RL, et al. Primary intradural extramedullary spinal mesenchymal chondrosarcoma: case report and literature review. BMC Musculoskelet Disord. 2019; 20: 408.
- Bae GS, Choi SW, Youm JY, Kim SH. Primary spinal dumbbell-shaped mesenchymal chondrosarcoma located intradurally and extradurally. J Korean Neurosurg Soc. 2011; 50: 468-71.
- Zhang Q, Xi Y, Li D, Yuan Z, Dong J. The utility of 18F-FDG PET and PET/CT in the diagnosis and staging of chondrosarcoma: a meta-analysis. J Orthop Surg Res. 2020; 15: 229.
- Kao YC, Lee JC, Huang HY. What is new about the molecular genetics in matrix-producing soft tissue tumors? -The contributions to pathogenetic understanding and diagnostic classification. Virchows Arch. 2020; 476: 121-134.
- Kilpatrick SE, Reith JD, Rubin B. Ewing Sarcoma and the History of Similar and Possibly Related Small Round Cell Tumors: From Whence Have We Come and Where are We Going? Adv Anat Pathol. 2018; 25: 314-326.
- Komal A, Riddle ND. Extraskeletal Mesenchymal Chondrosarcoma. Arch Pathol Lab Med. 2018; 142: 1421–1424.
- Wehrli BM, Huang W, De Crombrugghe B, Ayala AG, Czerniak B. Sox9, a master regulator of chondrogenesis, distinguishes mesenchymal chondrosarcoma from other small blue round cell tumors. Hum Pathol. 2003; 34: 263-9.
- Fanburg-Smith JC, Auerbach A, Marwaha JS, Wang Z, Santi M, Judkins AR, et al. Immunoprofile of mesenchymal chondrosarcoma: aberrant desmin and EMA expression, retention of INI1, and negative estrogen receptor in 22 female-predominant central nervous system and musculoskeletal cases. Ann Diagn Pathol. 2010; 14: 8- 14.
- Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours J Pathol. 2011; 224: 334-343.
- Fanburg-Smith JC, Auerbach A, Marwaha JS, Wang Z, Rushing EJ. Reappraisal of mesenchymal chondrosarcoma: novel morphologic observations of the hyaline cartilage and endochondral ossification and beta-catenin, Sox9, and osteocalcin immunostaining of 22 cases. Hum Pathol. 2010; 41: 653-662.