
Case Report
Austin J Clin Case Rep. 2024; 11(2): 1319.
Metabolic Epilepsy Associated with L-Serine Deficiency: Considerations Based on A Clinical Case
Calcîi C¹,²; Lavranciuc F¹*; Sprincean M¹,²; Gorincioi N³; Moldovanu M4; Marga S¹; Feghiu L³; Istratuc I²; Capestru E²; Revenco N¹,²; Groppa S¹?³; Hadjiu S¹,²
1Testemitanu State University of Medicine and Pharmacy, Chisinau, Republic of Moldova
2Mother and Child Institute, Chisinau, Republic of Moldova
3National Center of Epileptology, Chisinau, Republic of Moldova
4German Diagnostic Center, Chisinau, Republic of Moldova
*Corresponding author: Lavranciuc F Nicolae Testemitanu State University of Medicine and Pharmacy, Chisinau, Republic of Moldova. Email: lavranciucf@gmail.com
Received: March 13, 2024 Accepted: April 19, 2024 Published: April 26, 2024
Abstract
Purpose of Study: IEM are relatively rare causes of seizures in infants and children. L-serine deficiency that occurs in IEM is determined by a mutation in the genes encoding the enzymes involved in L-serine synthesis. The purpose of the study, through the example of a clinical case, was to emphasize the importance of metabolic screening evaluation of children with drug-resistant seizures for the fastest possible initiation of targeted treatment, thus preventing and minimizing the rate of complications.
Materials and methods: Presentation of a clinical case of a 4-year-old boy hospitalized a lot of times in the Neurology Department of our hospital due to polymorphic epileptic seizures, resistant to combined anticonvulsant treatment (Levetiracetam, Valproic Acid, Clonazepam), associated with cognitive and verbal decline, secondary to seizures. During the genetic testing for IEM, the c.777A>T mutation was detected in exon 7 of the PSAT1 gene – a gene involved in the coding of the PSAT enzyme that participates in the synthesis of L-serine, thus its severe deficiency is associated with the clinical picture of our patient.
Results: After the treatment with L-serine with a dose of 500 mg/kg/day divided into 3 doses, the child’s condition clinically improved. Seizures disappeared after 2 months of treatment with a positive cognitive and verbal dynamics.
Conclusions: Early diagnosis of seizures occurring in IEM is essential, as many IEMs are potentially treatable, and seizure control can only be achieved when they are properly treated.
Keywords: Seizures; L-serine; Inborn; Metabolic; Gene
Abbreviations: IEM: Inborn Errors of Metabolism; PSAT1: Phosphoglycerate Aminotransferase 1; PGDH: Phosphoglycerate Dehydrogenase; PSP: Phosphoserine Phosphatase; NMDA: N-Methyl-D-Aspartate; EEG: Electroencephalogram; MRI: Magnetic Resonance Imaging; FLAIR: Fluid Attenuated Inversion Recovery
Introduction
IEM are relatively rare causes of seizures in infants and children. Hereditary metabolic disorders include: amino acid metabolic disorders, energy metabolism disorders, cofactor-related metabolic disorders, purine and pyrimidine metabolic diseases, congenital glycosylation disorders, and lysosomal and peroxisomal disorders [1,2]. L-serine deficiency belongs to the category of hereditary metabolic disorders of amino acids. L-serine is a non-essential amino acid, but given the many important cellular functions of L-serine, it is considered a conditionally essential amino acid. L-serine is a precursor of important metabolites such as nucleotides, phospholipids and the neurotransmitters glycine and D-serine [3,4].
The genetic deficiency of L-serine is determined by a mutation in one of the three genes that encode the enzymes involved in L-serine biosynthesis: PGDH, PSAT1, PSP [5,6]. Children with congenital defects of the given enzymes show severe neurological abnormalities, such as: microcephaly, seizures refractory to treatment, neurodevelopmental delay, severe intellectual disability, growth deficiency, etc. Seizures occurring in L-serine deficiency are probably related to disturbances in NMDA receptor activation due to insufficient synthesis of glycine and D-serine [3,7]. The development of genetic technology has led to the identification of an increasing number of genes associated with epilepsy. Thus, Jie Wang et al. searching through several databases (OMIM, HGMD, and EpilepsyGene) and through recent publications on PubMed found 977 genes that are associated with epilepsy, of which 536 genes were classified as epilepsy-related genes. The deficiency of PSAT1, the enzyme involved in L-serine biosynthesis, was included in the list of 536 genes related to epilepsy [8].
Seizures occurring in metabolic disorders related to L-serine deficiency are refractory to anticonvulsants in almost all individuals, and seizures improve significantly after treatment with L-serine, thereby decreasing the need for chronic anticonvulsant medications. Improvement of EEG abnormalities may not occur until six months of L-serine treatment [7].
Case Report
A 4-year-old boy is admitted to the Department of Neurology of the Mother and Child Institute in the Republic of Moldova with absence and atonic seizures with head loss, mild verbal and cognitive retardation. These attacks started a year ago with generalized convulsive seizures, later the mother also noticed a cognitive and verbal decline in the child. The pregnancy was uneventful, the child was born naturally at 39 weeks. The birth proceeded without complications. The child was hospitalized multiple times in our ward for follow-up, and a series of investigations were carried out.
• EEG shows primary generalized complexes spike-slow waves and polyspikes-slow waves. Clinical events related to the recording were not observed (Figure 1).
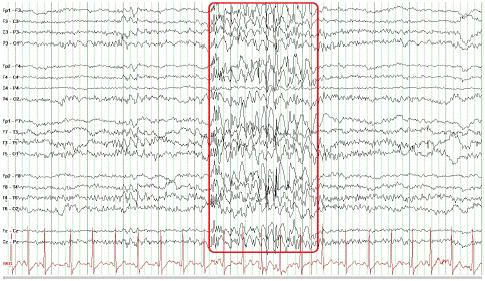
Figure 1: Electroencephalography of a 4-year-old child during sleep. It shows primary generalized spike-slow waves and polyspikes-slow waves. Clinical events related to the recording were not observed.
• Brain MRI, FLAIR sequence - axial, sagittal and coronal planes. Gliotic foci in the periventricular white matter of the frontal lobes on both sides (Figure 2).
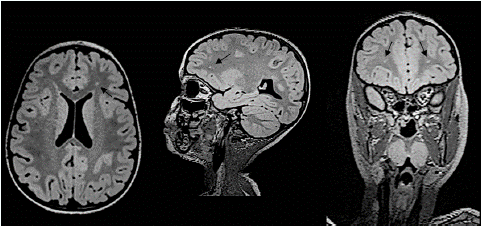
Figure 2: Brain MRI, FLAIR sequence - axial, sagittal and coronal planes. Gliotic foci in the periventricular white matter of the frontal lobes on both sides.
• The psychologist's consultation demonstrated a cognitive decline according to the Wechsler scale of 65.
• Eredocollateral anamnesis: There are no cases of seizures or known genetic diseases in the family.
Initially, the child was on antiepileptic treatment with Valproic Acid at a dose of 30 mg/kg, but considering that the seizures were repeated despite the background of antiepileptic treatment, Levetiracetam was associated with a dose of 40 mg/kg. Because the child continues to have seizures despite the background of antiepileptic treatment administered, he was consulted by a geneticist to rule out a genetic cause of epilepsy. Meanwhile, Clonazepam with a dose of 0.05 mg/kg was added to the treatment. The geneticist recommended the Epilepsy panel where it was determined:
• The c777A>T mutation in exon 7 of the PSAT1 gene with autosomal dominant transmission, being associated with PSAT deficiency (Table 1).
![]()
Gene: PSAT1Exon: 7
Locus: chr9:8093262
Mutation: c.777A>T
Transmission: AD/AR
The type of mutation: SNV
Genotype: A/T
Reference genotype: T/T
Amino acid: Lys259Asn
Classification ClinVar: Uncertain
Diseases associated with the gene according to OMIM: Phosphoserine aminotransferase deficiency; Neu-Laxova syndrome 2.
Table 1: The result of the genetic testing for Epilepsy. Mutation c.777A>T in exon 7 of the PSAT1 gene.
Following the genetic results, treatment with L-serine 500 mg/kg/day divided into 3 doses throughout the day was indicated. On the background of treatment with L-serine, the child's condition clinically improved. The seizures became less frequent in the first month of treatment, with their total disappearance after 2 months of treatment, and from a cognitive and verbal point of view, a positive dynamics is attested, with the increase of the score on the Wechsler scale up to 80.
Discussions
Serine deficiency disorders include a spectrum of diseases ranging from the fatal prenatal-onset Neu-Laxova syndrome to infantile, juvenile, or adult-onset serine deficiency. Neu-Laxova syndrome is characterized by severe intrauterine growth deficiency, microcephaly, congenital bilateral cataracts, characteristic dysmorphic features, limb abnormalities, and ichthyosis [9].
Infants are usually stillborn or die in early childhood. Infantile-onset serine deficiency is characterized by seizures, microcephaly, developmental delay, intellectual disability, and spastic tetraplegia. People who develop serine deficiency in their early teens have seizures, and many develop spastic tetraplegia. Serine deficiency in adults is characterized by progressive axonal polyneuropathy with ataxia and possible cognitive impairment [10,11].
The child presented above was diagnosed with the infantile form of serine deficiency, which presented with convulsions resistant to combined antiepileptic treatment and verbal and cognitive retardation. In order to exclude a genetic cause of epilepsy, the child was genetically investigated at the Epilepsy panel, where the c777A>T mutation was detected in exon 7 of the PSAT1 gene - the gene involved in L-serine synthesis, thus the severe deficiency of it is associated with the clinical picture mentioned above.
Seizures caused by serine deficiency are rare. In the case of diagnosis and early initiation of substitution treatment, a positive dynamics is observed in the control of seizures by decreasing the frequency and intensity of seizures even with their total disappearance [7,12].
On the background of treatment with L-serine at a dose of 500 mg/kg/day divided into 3 doses, the child's condition clinically improved both on account of the decrease in frequency and intensity of seizures until they disappearance, and from a verbal and cognitive point of view the evolution was with positive dynamics. According to the studies carried out, seizures improve significantly after treatment with L-serine, thus reducing the need for chronic anticonvulsant drugs, and after a few months the electroencephalographic track also normalizes [7].
Large doses of L-serine are required to correct serine deficiency and to achieve serine values within reference limits in plasma and cerebrospinal fluid. L-serine therapy is usually started at a dose of 200-400 mg/kg/day (administered orally and divided into 4-6 doses), as myoclonus has been observed in some individuals at higher doses. The dose of L-serine is gradually increased to 500-700 mg/kg/day in 4 doses. At the same time, a dose of 400 mg/kg/day is considered insufficient to prevent the recurrence of seizures [9].
Brassier and colleagues describe two clinical cases with genetic L-serine deficiency that underwent L-serine replacement treatment. In the first case, a child (aged 4.5 years) who presented seizures resistant to combined antiepileptic medication (Valproic Acid, Clonazepam, Topiramate and Levetiracetam) is reported. The initiation of substitution therapy with L-serine significantly reduced the frequency and intensity of attacks (up to 50%) with a dose of 500 mg/kg/day in 4 doses, and once with an increase in the dose up to 650 mg/kg/day in 4 doses it led to the disappearance of seizures with the gradual cessation of Topiramate and Clonazepam. In the case of patient 2 (child aged 3 months), the administration of L-serine significantly improved spasticity at the initial dose of 400 mg/kg/day in 4 doses, but despite the fact that the dose of L-serine was increased up to 950 mg/kg/day in 4 doses, the child was left with serious neurological sequelae, remaining immobilized in bed [13].
S. N. van der Crabben and colleagues emphasize the importance of selecting the correct doses of L-serine through the prism of a clinical case where they report a child diagnosed with a genetic deficiency of L-serine and seizures who started treatment with L-serine with a dose of 500 mg/kg /day divided into 4 doses, later with its reduction to 400 mg/kg/day due to some suspected adverse reactions. After 7 months on the background of this small dose, the child developed West syndrome with infantile spasms and hypsarrhythmia on EEG. The dose of L-serine was increased to 600 mg/kg/day and later to 700 mg/kg/day with subsequent normalization of the EEGs [14] .
At the moment, the child from our case is under neurological supervision for 1 year with neurocognitive, imaging and electrophysiological evaluation.
Conclusions
In the context of the discussed clinical case, we suggest that IEMs should always be considered when evaluating infants and young children presenting with drug-resistant seizure episodes. These conditions (IEMs) may require specific therapies, and proper management of the underlying etiology can help control seizures.
Genetic testing should be prioritized, as it offers the highest diagnostic yield in children with pharmacoresistant epilepsy.
Early diagnosis of the etiology of epileptic seizures is crucial, as timely intiation of appropriate treatment can contribute to improving overall developmental delays.
References
- Dulac O, Plecko B, Gataullina S, Wolf NI. Occasional seizures, epilepsy, and inborn errors of metabolism. Lancet Neurol. 2014; 13: 727-39.
- Sharma S, Prasad AN. Inborn Errors of Metabolism and Epilepsy: Current Understanding, Diagnosis, and Treatment Approaches. Int J Mol Sci. 2017; 18: 1384.
- Tabatabaie L, Klomp LW, Berger R, Koning TJ. L-serin synthesis in the central nervous system: a review on serine deficiency disorders. Mol Genet Metab. 2010; 99: 256–62.
- Almannai M, Mahmoud RAA, Mekki M, Wl-Hattab AW. Metabolic Seizures. Front Neurol. 2021; 12: 640371.
- Shen Y, Peng Y, Huang P, Zheng Y, Li S, Jiang K, et al. Juvenile-onset PSAT1-related neuropathy: A milder phenotype of serine deficiency disorder. Front Genet. 2022; 13: 949038.
- Koning TJ, Klomp LWJ. Serine-deficiency syndromes. Curr Opin Neurol. 2004; 12: 197-204.
- Crabben SN, Koning TJ. Serine deficiency disorders; tratable causes of seizures. Journal of Pediatric Epilepsy. 2014; 3: 241–8.
- Wang J, Lin Z-J, Liu L, Xu H-Q, Shi Y-W, Yi Y-H, et al. Epilepsy-associated genes. Seizure. 2017; 44: 11-20.
- Acuna-Hidalgo R, Schanze D, Kariminejad A, Nordgren A, Kariminejad MH, Conner P, et al. Neu-Laxova Syndrome Is a Heterogeneous Metabolic Disorder Caused by Defects in Enzymes of the L-Serine Biosynthesis Pathway. Am J Hum Genet. 2014; 95: 285–93.
- Fons-Estupiña MC. Síndromes epilépticos de inicio neonatal. Etiologías y proceso diagnóstico / Neonatal onset of epileptic syndromes. Causations and diagnostic process. REV NEUROL. 2018; 66.
- Benke PJ, Hidalgo RJ, Braffman BH, Jans J, Gassen KLI, Sunbul R, et al. Infantile Serine Biosynthesis Defect Due to Phosphoglycerate Dehydrogenase Deficiency: Variability in Phenotype and Treatment Response, Novel Mutations, and Diagnostic Challenges. J Child Neurol. 2017; 32: 543-549.
- Brassier A, Valayannopoulos V, Bahi-Buisson N, Elsa Wiame LH, Boddaert N, Kaminska A, et al. Two new cases of serine deficiency disorders treated with l-serine. Eur J Paediatr Neurol. 2016; 20: 53–60.
- Crabben SN, Koning TJ, Adam MP, Feldman J, Mirzaa GM, Pagon RA, et al. Serine Deficiency Disorders. Gene Reviews. 2023.
- Crabben SN, Verhoeven-Duif NM, Brilstra EH, Maldergem L, Coskun T, Rubio-Gozalbo E, et al. An update on serine deficiency disorders. J Inherit Metab Dis. 2013; 36: 613–9.