
Case Report
Austin J Clin Case Rep. 2024; 11(3): 1325.
Watch out For Skin Discoloration; Cyanotic Extremities Turned Purpura Fulminans; An Unusual Presentation
Agogbuo O1; Undavalli C2; Kanuri S2*; Diala C3; Elhalwagi B4; Frase L5; Vegi Y6
1Internal Medicine Residency, CHRISTUS Good Shepherd/ Texas A&M University School of Medicine.
2University of Texas Health Science Center, Tyler, TX USA
3Internal Medicine Residency, Piedmont Athens Region Medical Center, Athens, Georgia
4Faculty, Christus Health, Pulmonology- Critical Care Department, Christus Good Shepherd Hospital, Longview, TX USA
5Texas US Oncology, Longview TX, USA
6Internal Medicine, CHRISTUS Trinity Clinic, Longview, TX USA
*Corresponding author: Sri Harsha Kanuri, MD, PhD MPH, Research Fellow, Biomedical Research Center, 3900 University Blvd, UT Tyler School of Medicine, Tyler, TX 75799, USA. Email: harsha9009@gmail.com
Received: June 03, 2024 Accepted: July 01, 2024 Published: July 05, 2024
Abstract
Purpura Fulminans (PF) is a dermatological disorder characterized by manifestation of purpuric lesions, ecchymosis, and skin necrosis. It is commonly associated with sepsis, Disseminated Intravascular Coagulation (DIC), microvascular thrombosis and circulatory collapse. It has been documented in the neonates, children, and adults. It is mainly classified into three types namely genetic, idiopathic, and infectious forms. The main pathophysiology is dwindling of anticoagulant factors with predomination of coagulant factors in the blood circulation, thence increasing the penchant for hypercoagulation tendency in the blood circulation. This will lead to formation of microthrombi in the dermal vasculature, thus providing deep-rooted basis for inception of skin lesions usually seen in this clinical entity. Unfortunately, due to lack of therapeutic modalities, there is high mortality and morbidity in these patients. We present a clinical case of healthy and asymptomatic women with incidental auto-infarction of spleen, who presented with streptococcal pneumonia, and septic shock. Her hospital course was complicated with PF and disseminated intravascular coagulation. She initially developed painful purpuric lesions which slowly progressed to non-blanching purpuric ecchymosis and flaccid bullae. These lesions were associated with cyanosis of the nose, lips and feet, and vascular thrombosis Streptococcal pneumonia was the elusive trigger which provoked the sepsis induced destruction and depletion of anti-coagulant factors, hence provoking the inception of PF. This is one the rare cases of PF, where the patient demonstrated excellent prognosis, impressive clinical recovery, and no clinical sequelae with antibiotics, IVIG, anticoagulation and supportive therapy.
Keywords: Purpura fulminans; Erythematous nodules; Cyanosis; DIC; Bacterial infections; Asplenia; Sepsis
Introduction
Purpura Fulminans (PF) is a hematological emergency manifesting as skin necrosis and disseminated intravascular coagulopathy, (DIC) [1]. It progresses to multiorgan failure due to thrombotic occlusion of medium and small-sized blood vessels [2]. It may occur as an autoimmune response to otherwise benign childhood infections or severe sepsis or as presenting symptoms of severe inheritable deficiency of protein C and S [3]. Infectious PF has a propensity to present as DIC due to diffuse intravascular thrombosis and hemorrhagic infarction of the skin [1,4]. It presents in infancy, childhood, or adulthood as ecchymosis, skin lesions, fever, and hypotension [4]. It is commonly seen as a complication of sepsis related to Neisseria, Strep pneumonia and H. influenzae [1]. Despite aggressive fluid resuscitation and antibiotic management, sepsis induced PF carries a mortality rate of 3-36% [5]. Other types of PF (Genetic & Idiopathic) have varied mortality depending upon the age of presentation, and risk factor profile. We present the case of PF secondary to streptococcal pneumonia sepsis in a patient with an incidental finding of auto infarction of the spleen in an otherwise healthy and asymptomatic patient. In our case, an immunocompetent woman in her 40s with incidental asplenia/auto-infarct spleen presented with septic shock from strep pneumonia bacteremia. Her hospital course rapidly progressed to Disseminated Intravascular Coagulopathy (DIC) and PF aggressively treated with broad-spectrum antibiotics, and IVIG infusions. Our patient had an initial presentation raising concern for methemoglobinemia, carbon monoxide poisoning and sepsis as possible differential diagnoses. The source of her strep pneumonia bacteremia remained an elusive trigger.
Case Report
A 45-year-old white female presented to the emergency department complaining of subjective shortness of breath that started 1 hour before presentation, subjective fever, fatigue, generalized weakness, diarrhea, vomiting, and nausea that started 2 days prior to arrival. Patient was not a known user of tobacco or alcohol. No prior surgical history, hematological disorders or recent travel history was noted. Upon evaluation in the Emergency Room [ER] visit, she was found to have a temperature of 102F, hypotensive with systolic blood pressure in the 70s, and consequently received 3 L of bolus fluids. Other vital signs were stable. On physical examination, she is noted to have mottling of bilateral lower extremities. Complete blood count revealed leukocytosis with a total WBC count of 11.7 cells per mL, along with COVID PCR negative, and lactic acid of 2.6. Despite 4 L of fluid resuscitation, she developed persistent hypotension, borderline tachycardia [HR- 110-120 bpm], tachypnea, acute hypoxic respiratory failure, and septic shock. The patient was admitted to ICU. Blood cultures were obtained before starting broad-spectrum antibiotic coverage with intravenous vancomycin and Zosyn. Upon looking into patient’s coagulation profile, PT & PTT were prolonged and fibrin degradation products were grossly elevated suggesting DIC. Infectious work-up was remarkable for blood cultures that grew streptococcus pneumonia. Within the next 48 hours, the patient developed a diffusely painful petechial rash which progressed from non-blanching purpuric ecchymosis lesions to flaccid bullae resulting in skin sloughing especially in the palms of the upper extremities, cyanosis of nose feet lips, with pain. CT scan of the abdomen-pelvis was done in the evaluation of possible source of bacteremia showed auto infarction of spleen/questionable asplenia. The patient received a 14-day course of broad-spectrum antibiotics, heparin drip, and IV immunoglobulin. Protein C concentrate was not available, hematology recommended starting on 2 units FFP every 6 hours. Aggressive management protocol including specific antimicrobial cocktail, maintaining vascular perfusion, treatment of the underlying cause, and importantly removing the stimulus for coagulation tendencies leading to purpura was carefully considered in this patient. The patient responded well to therapy with complete resolution of dermatological manifestations and associated clinical symptoms. This is one of the rare cases of purpura fulminans secondary to asplenic sepsis which showcased an excellent prognosis, impressive clinical recovery, and absent clinical sequelae. Careful clinical follow-up and prophylactic antibiotic therapy is recommended given the asplenia in this patient to prevent further bacterial infections and associated complications.
Discussion
Since it being discovered in 1884, the incidence and prevalence of Purpura Fulminans (PF) varies according to its subtype [6]. The prevalence of hereditary form and acute infectious form of purpura fulminans are 1 case per 1000,000 live births and 1 case per 100,000 live births respectively [1,7]. Most commonly, it is seen in neonatal patients with hereditary homozygous protein C deficiency, but older patients presenting with a heterozygous protein C deficiency is not uncommon. PF may also occur in settings of severe acquired protein C deficiency as may occur in patients with severe bacterial infections. PF is a severe complication of meningococcal disease and it has been documented in roughly 10-20% of the cases of meningococcal sepsis [8]. In severe cases, the case mortality rate of purpura fulminans is as high as 50% [9]. In children presenting with idiopathic PF, there might underlying protein C deficiency, Factor V mutations and prothrombin mutations in 4%, 16% and 12% of the cases respectively [10]. In a French multicenter retrospective study, it was assessed that the prevalence of inherited thrombophilia or antiphospholipid antibodies in the patients with idiopathic PF was approximately 28% [10].
Although sepsis and its complications are common causes of PF, other possible causes should be evaluated including acquired protein C or Protein S deficiency, antithrombin III deficiency, other genetic mutations affecting coagulation, underlying meningococcal or streptococcal infections, and certain medications or therapy such as warfarin, Heparin or chemotherapy [1,1]. The disease process may be exacerbated by unrecognized underlying factors, which can result in a worse outcome with associated high morbidity and mortality. There are three types of purpuras namely, idiopathic, acute infectious and neonatal [1].
In the idiopathic form, the dawning of purpura usually follows recent viral infection (Varicella Zoster Virus [VZV] & Human herpe virus 6 [HHV6]) [12]. These viral infections will instigate the generation of auto-antibodies, which can possibly interact with anticoagulant factors, henceforth eliciting their undersupply in the systemic circulation[12]. Anti-protein-S antibodies were witnessed in 50% of idiopathic PF cases, thus supporting the speculation of autoimmune origin for their inception [12]. In immunocompetent patients, these infections are short-lived due to their proficient immune system, with the majority of them having an uneventful recovery with no long-term sequelae. In the contrary, in susceptible individuals, overactive immune system retaliates by procreating antibodies against these pathogens [12,13]. A prejudicious repercussion of this culmination is that these antibodies develop cross reactivity with protein-S alone, or protein-C & antithrombin III factors [12,13]. This will incite their binding, destruction, and consequent excretion of these anticoagulant factors, thereby enkindling their transient deficiency in the blood [1,12,13]. Protein S is a vitamin dependent protease that mainly functions as a cofactor for activated protein C [APC], resultantly inhibiting clotting mechanism via restraining the activity of clotting factors Va and VIIIa [14]. On grounds of this, protein-S factor desecration alone initially is sufficient to downgrade and inactivate Protein-C mediated pathway, thus creating a fertile bed for transpiration of thrombotic tendencies in these susceptible infants.
Infections such as Neisseria meningitidis, Capnocytophaga, Streptococcus pneumoniae, Streptococcus pyogenes, Staphylococcus aureus, and Haemophilus influenzae are implicated for of causation of PF in the asplenic patients [15]. Additionally, bacterial species (Nesisseria Meningitidis, Salmonella typhi, Proteus mirabilis, Escherichia coli, Enterobacter species, Streptococcus pyogenes, Streptococcus agalactiae. Leptospira and Klebsiella penumoniae) fungi (Candida, Aspergillus, Fusarium, & Cryptococcus neoformans) protozoa (Plasmodium viva and Plasmodium falciparum), and viruses (West Nile Virus, & Varicella Zoster virus) are also responsible for triggering the disease pathology [4]. The endotoxin released from these pathogens will have a pernicious effect on the circulating anticoagulant factors (protein-S, protein-C & antithrombin III factors) by precipitating their lysis, destruction and elimination [16,17]. For instance, in meningococcal septicemia, the release of bacterial endotoxin triggers the annihilation of anti-coagulant agents via amplifying the production of pro-inflammatory cytokines such as IL-1, IL-12, interferon-7 & tumor necrosis factor-alpha [2,18]. Eventually, this will provoke their purging, and stonewall them from enforcing their anti-coagulant effect in the blood circulation.
The neonatal form usually presents within 5-7 days following birth and these infants have inherent genetic deficiency of protein-C, protein-S and antithrombin-III [1,4,19-22]. In light of their relative absence, a paralyzing effect on the anti-coagulant arm of the coagulant system will be the ultimate ripple-effect, thence engendering a opportune occasion for hegemony of the pro-coagulant arm. This will procreate a state of affairs where there is an amplified proclivity to develop widespread thrombotic tendency, henceforth making infants manifest with DIC and ensuing after-effects.
Evidently, in all the three forms of purpura fulminans, decrease or disintegration of the vital anti-coagulant agents brings to pass a grievous set of circumstances, where there is a predominance of pro-coagulant factors along with concurrent deactivation of anti-coagulant factors [23]. In the physiological state, towering presence of these anti-coagulant factors will exert a negative effect on the activity of pro-coagulant factors (Va & VIIIa), as a result curbing the thrombin synthesis and subsequent fibrogenic pathways [24,25]. This will prevent the development of thrombosis and ensures continuous blood supply to the systemic tissues [24,25]. Unfortunately, the ominous turn of events during various forms of purpura fulminans will spark off an unhampered activity of pro-coagulant factors (Va & VIIIa), thence amplifying the thrombin synthesis and thrombotic tendency seen commonly in these patients [2,23]. These downstream signaling events form the deep-seated basis for widespread manifestation of microvasculature thrombosis, disseminated intravascular coagulation, hemorrhage and circulatory collapse witnessed in these patients [2]. The pathological findings present in the purpura fulminans regardless of its type include dermal vascular thrombosis and secondary hemorrhagic necrosis [18]. With that being said, these aforementioned pathological changes are responsible for inception of purpura, ecchymosis, bullae and necrosis seen in these patients.
The timing of the clinical presentation will ultimately depend upon the type of purpura fulminans. In neonatal form, the clinical presentation will be within 5-10 after birth [1,26]. However, in the idiopathic form, there will be a lag period of days to weeks after an antecedent bacterial or viral infection [1,27]. In most instances, the prior infection is very mild and has subsided completely before the onset of dermatological symptoms, with majority of them having no recollection of this antecedent illness [1]. In the contrary, the dawning of clinical symptoms will coincide with acute severe bacterial or viral infection [1,16]. Idiopathic PF can be differentiated from sepsis induced PF by the predominance of microthrombi in the dermal vasculature rather than systemic vasculature [28].
In our present case, the patient presented initially with a diffusely painful petechial rash which then progressed to non-blanching purpuric ecchymosis lesions and flaccid bullae (Figure 1). These changes resulted in skin sloughing especially in the palms of the upper extremities, along with cyanosis of nose, feet, and lips, with associated pain. In most cases, there will be signs of cutaneous hemorrhage and necrosis secondary to culmination of vascular thrombosis and DIC, all of which have been documented in our patient. Cutaneous pain followed by erythema and petechiae was also seen in our patient. In typical case of purpura fulminans, dark red-purple lesions tend to appear in the distal extremities, and these are non-blanchable despite applying pressure [4,19]. Prompt diagnosis and therapeutic management is necessary to curb the disease progression. Delay in diagnosis and therapy incites unhindered disease progression with subsequent appearance of painful indurated well-demarcated purple papules with erythematous borders [4,19]. Untreated, these lesions convert into erythematous petechiae, necrotic foci associated with the formation of bullae and vesicles [4,19]. In the final stages, spread of disease to the subcutaneous tissues including muscle and bone results in gangrenous necrosis and this usually effects distal extremities with a tendency to spread inwards in a retrograde fashion [4,19].
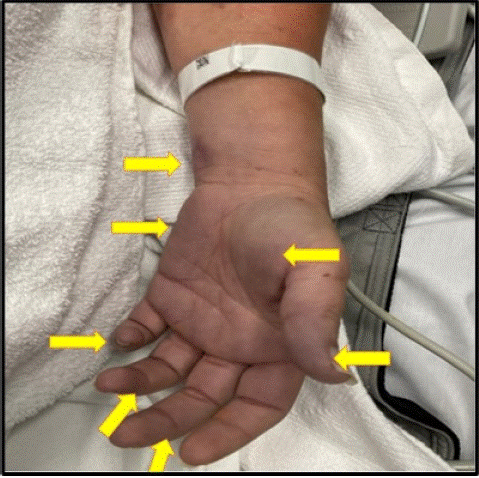
Figure 1: Skin Lesions in our PF. Our patient was presented with a diffusely painful petechial rash which then progressed to non-blanching purpuric ecchymosis lesions (Arrows). These changes were associated with flaccid bullae which later progressed into skin sloughing especially in the palms of the upper extremities.
Some of the important differential diagnosis that should be considered in case of purpura fulminans include Vasculitis, Thrombophlebitis, Heparin induced thrombocytopenia, Thrombotic thrombocytopenic purpura, Henoch Schonlein purpura, Antiphospholipid syndrome, Toxic shock syndrome, Warfarin induced necrosis, Necrotizing fasciitis, Meningococcemia and Calciphypaxis [1,4,27]. As the clinical spectrum and severity of purpura fulminans is directly contingent upon its subtype, efforts should be directed towards uncovering the underlying cause by diligently pursuing pertinent investigations. This might involve a cocktail of lab tests including blood cultures, genetic testing, coagulation parameters (low protein C&S, increased PT, increased aPTT, low fibrinogen, elevated fibrin degradation products & elevated D-dimers), liver function tests, Basic metabolic profile and CBC & differential [1,4,29,30]. The management of purpura fulminans mainly entails uncovering the underlying cause and treating it aggressively. In moderate to severe cases, in-patient admission is deemed necessary for providing supportive care, fluids (inotropic support), correction of acid-base disturbances and rectification of electrolyte abnormalities [7]. Due to their increased predisposition to develop widespread thrombosis, anticoagulation with heparin or warfarin should be considered in case-by-case basis [28]. The clinical efficacy of anticoagulants in preventing the development of thrombosis and averting the imbalance of coagulation factors might afford a therapeutic benefit of intercepting the materialization of dermal necrosis and ensuing clinical manifestations of purpura fulminans [28]. Surgical intervention including the wound debridement, fasciotomies, escharotomies and rarely amputation might be possibly needed on a case-by-case basis based on the involvement of underlying muscle/bone and presence of compromised tissue perfusion [31]. In Purpura fulminans secondary to acute infectious disease, starting broad spectrum antibiotic administration with vancomycin + beta-lactum inhibitor + clindamycin while waiting for results of blood cultures would be a prudent strategy [1]. Keeping in mind the vulnerability to develop serious life-threatening infections in asplenic patients, it would be prudent to consider a Pill-Pocket strategy in these specific set of patients [15,32]. This encompasses susceptible patients self-administer antimicrobials on the first day of developing fever or any sign signifying infection [15,32]. This will decrease their propensity to develop fatal bacterial infections, thus their ensuing risk of developing purpura fulminans and DIC [15,32]. This prophylactic strategy might potentially lessen the morbidity and mortality in these susceptible patients [15,32-34].
In PF cases where autoimmunity is suspected, clinical management should be focused on blunting the production of autoantibodies so that they will be lesser chance of their binding to the anti-coagulant factors, a transpiration that is tied to their inactivation and elimination from the blood. In this regard, few clinical studies had showcased a therapeutic benefit with the utilization of corticosteroids, whose clinical efficacy might relate to their immunosuppressive, and anti-inflammatory properties [18,35]. Owing to the fact that, toxins released by staphylococcal bacteria might be responsible for instigating the procreation of autoantibodies, any interventions aimed to nullify these toxins with IVIG can be beneficial in reversing the disease pathology in PF[1] [36,38]. In some cases, plasma exchange might be moderately beneficial by purging pro-inflammatory factors (IL-6 and TNFalpha), bacterial toxins and autoantibodies [7,39].
Microthrombi provoked poor tissue perfusion will trigger release of prostaglandin, which later arouses A delta and C nerves, thence furnishing pathophysiological basis for emergence of pain in the purpura fulminans [28,40,41]. This pain in PF can be partly intercepted by topical nitroglycerin and epoprostenol (Prostacyclin), which are mainly efficacious by triggering vasodilation and curbing platelet aggregation [40-42]. Replenishing clotting factors like protein C and antithrombin III might afford clinical benefit by buttressing the anticoagulant arm as well as by restoring the balance in the coagulation system [43-45]. Recombinant tissue plasminogen activator (rTPA) might be beneficial in PF with its clinical efficacy upspringing from its proficiency in energizing clot-specific fibrinolysis, henceforth triggering disruption of fibrin clots in the blood circulation [46,47]. Plasmapheresis is one of the therapeutic options in PF due to ability to filter out endotoxins, toxins, pro-inflammatory mediators, cytokines in addition to purging out excessive fluid [28]. Furthermore, fresh frozen plasma and cryoprecipitate has been tried and they act by restoring fibrinogen levels and reversing the PT and aPTT levels [11,28]. Protein C concentrate replenishment has been used previously with some clinical trials, case-series and case-controls studies in acquired or genetic protein C deficiency, with some demonstrating no clinical benefit while showing modest clinical benefit [48-52]. In patients with sepsis induced PF and vascular insufficiency, epidural sympathetic block might be considered in addition to supportive measures as it tends to restore the skin perfusion and preserve tissue viability of lower extremities, resultantly preventing necrosis, gangrene, and amputation in severe cases [53,54].
In some instances, clinicians had found modest success with usage of Leech Saliva which harbors Hirudin, a potent inhibitor of thrombin. By its ability to invade and bind to thrombin, it can be clinically efficacious in regressing clots associated with thrombin [55]. As a supplementary treatment, intravenous dextran has been tried and its clinical benefit in PF springs from its ability to coat the vessel wall, RBC & platelets, repress cell aggregation and suppress blood thickening, henceforth affording modest symptomatic relief especially when co-administered with other therapeutic interventions in PF [28].
Conclusion
Purpura fulminans is a rare clinical diagnosis that should be promptly recognized and empirically treated especially with signs of sepsis and DIC. Our case demonstrated a rare cause of incidental asplenia with questionable auto infarct associated with streptococcal pneumonia as a cause of purpura fulminans. Our patient had a dramatic clinical response where there was a complete resolution of dermatological manifestations and clinical symptoms with supportive therapy including antibiotics, anticoagulants, fresh frozen plasma, and immunoglobulins. Given the asplenia and risk of vulnerability to further infections, recommendation of prophylactic antibiotics and pill-pocket cocktail would be an efficient therapeutic strategy to avert infection provoked PF in these subsets of patients. Early recognition and treatment of PF is essential to reduce mortality and prevent long-term health sequela.
Learning Points
1. Purpura fulminans is a skin disorder characterized by clinical spectrum ranging from erythema, petechia, purple papules, vesicles, bullae to gangrenous necrosis.
2. It is mainly classified into three subtypes namely neonatal, acute infectious and idopathic
3. The underlying pathophysiology encompasses plummeting anticoagulant factors in combination with burgeoning pro-coagulant factors.
4. Incidental asplenia due to auto infarction can eventually amplify the risk of developing severe bacterial infections including pneumococcal or staphylococcal infections, a ramification that would incite thrombotic episodes, DIC and purpura fulminans.
5. Management is primarily dependent upon treating the underlying cause along with supportive management to replenish the anti-coagulant factors.
6. Due to the propensity to develop widespread thrombosis, it would be prudent to include anticoagulants in the treatment regimen.
7. In cases where autoimmunity is suspected, usage of corticosteroids to blunt the production of autoantibodies should be necessary to afford modest therapeutic benefit.
Author Statements
Author’s Contributions
Conceptualization, S.H.K, A.O.; Methodology, N.A.; Software, N.A.; Validation, N.A; Formal Analysis, N.A.; Investigation, N.A. Resources, N.A.; Data Curation, N.A.; Writing– Original Draft Preparation, S.H.K & A.O. .; Writing– Review & Editing, S.H.K., A.O. U.C., D.C., E.B., F.L. & Y.V.; Visualization, S.H.K.; Supervision, U.C., D.C., E.B., F.L. & Y.V.; Project Administration, N.A; Funding Acquisition, N.A.
References
- Perera TB, M LH, Purpura Fulminans. Stat Pearls. 2024.
- Morales Hernandez MDM, M Carranza, B Patel, J Calvert, G Masri. Purpura Fulminans in a Patient with Septic Shock due to Escherichia coli Bacteremia with Emphysematous Pyelitis. Cureus. 2021; 13: e13249.
- Yuen P, Cheung A, Lin HJ, Ho F, Mimuro J, Yoshida N, et al. Purpura fulminans in a Chinese boy with congenital protein C deficiency. Pediatrics. 1986; 77: 670-6.
- Shenoy R, Nanjappa S, Eaton K, Prieto-Granada C, Messina JL, Greene J. Purpura Fulminans: A Case Report and Review of All Causes. Infectious Diseases in Clinical Practice. 2017: 25.
- Madhi F, Ouldali N, Levy C, Taha MK, Cohen R, French Pediatric Meningitis Network. Factors associated with death in children with purpura fulminans: a French national prospective cohort study. Critical Care. 2021; 25: 181.
- Ruiz de Villa A, K Charles, P Okonoboh. A Rare Case of Purpura Fulminans in the Setting of Klebsiella pneumoniae Bacteremia. Cureus. 2022; 14: e22921.
- Kim MC, J Patel. Recognition and Management of Acute Purpura Fulminans: A Case Report of a Complication of Neisseria meningitidis Bacteremia. Cureus. 2021; 13: e13704.
- Bleeker-Rovers CP, HJC De Vries. 13 - Dermatologic Manifestations of Systemic Infections, in Infectious Diseases (Fourth Edition), J. Cohen, W.G. Powderly, and S.M. Opal, Editors. Elsevier. 2017; 113-121.e1.
- Harrison LH, DM Granoff, AJ Pollard. 38 - Meningococcal Capsular Group A, C, W, and Y Conjugate Vaccines, in Plotkin's Vaccines (Seventh Edition), S.A. Plotkin, et al., Editors. Elsevier. 2018; 619-643.e11.
- Theron A, Ayadi S, Boissier E, Dautremay O, Schved JF, Sirvent N, et al. Post-viral idiopathic purpura fulminans is associated with inherited thrombophilia and anti-cardiolipin antibodies. Frontiers in Pediatrics. 2023; 11: 1197795.
- Talwar A, S Kumar, MG Gopal, AS Nandini. Spectrum of purpura fulminans: report of three classical prototypes and review of management strategies. Indian J Dermatol Venereol Leprol. 2012; 78: 228.
- Theron A, Dautremay O, Boissier E, Zerroukhi A, Baleine J, Moulis L, et al. Idiopathic purpura fulminans associated with anti-protein S antibodies in children: a multicenter case series and systematic review. Blood Adv. 2022; 6: 495-502.
- Samman K, CK Le, B Michon. An Atypical Case of Idiopathic Purpura Fulminans. Journal of Pediatric Hematology/Oncology. 2022; 44: 479-481.
- Gupta A, TA, Gupta K, et al. Protein S Deficiency. Treasure Island (FL) StatPearls Publishing: StatPearls Publishing. 2024.
- Hale AJ, M LaSalvia JE, Kirby A Kimball, R Baden. Fatal purpura fulminans and Waterhouse-Friderichsen syndrome from fulminant Streptococcus pneumoniae sepsis in an asplenic young adult. ID Cases. 2016; 6: 1-4.
- Betrosian AP, T Berlet, B Agarwal. Purpura fulminans in sepsis. Am J Med Sci. 2006; 332: 339-45.
- Hesselvik JF, J Malm, B Dahlbäck, M Blombäck, Protein C, protein S and C4b-binding protein in severe infection and septic shock. Thromb Haemost. 1991; 65: 126-9.
- Darmstadt GL. Acute infectious purpura fulminans: pathogenesis and medical management. Pediatr Dermatol. 1998; 15: 169-83.
- Francis RB, Jr. Acquired purpura fulminans. Semin Thromb Hemost. 1990. 16: 310-25.
- Tuddenham EG, Takase T, Thomas AE, Awidi AS, Madanat FF, Hajir MMA, et al. Homozygous protein C deficiency with delayed onset of symptoms at 7 to 10 months. Thromb Res. 1989; 53: 475-84.
- Limperger V, Klostermeier UC, Kenet G, Holzhauer S, Gelas MA, Finckh Y, et al. Clinical and laboratory characteristics of children with venous thromboembolism and protein C-deficiency: an observational Israeli-German cohort study. Br J Haematol. 2014; 167: 385-93.
- Branson HE, J Katz, R Marble, JH Griffin. Inherited protein C deficiency and coumarin-responsive chronic relapsing purpura fulminans in a newborn infant. Lancet. 1983; 2: 1165-8.
- Colling ME, PK Bendapudi. Purpura Fulminans: Mechanism and Management of Dysregulated Hemostasis. Transfusion Medicine Reviews. 2018; 32: 69-76.
- Bertina RM. The role of procoagulants and anticoagulants in the development of venous thromboembolism. Thromb Res. 2009; 123: S41-5.
- Dahlback B. Blood coagulation and its regulation by anticoagulant pathways: genetic pathogenesis of bleeding and thrombotic diseases. Journal of Internal Medicine. 2005; 257: 209-223.
- Irfan Kazi SG, Siddiqui E, Habib I, Tabassum S, Afzal B, Khan IQ. Neonatal Purpura Fulminans, a rare genetic disorder due to protein C deficiency: A case report. J Pak Med Assoc. 2018; 68: 463-465.
- Ghosh SK, D Bandyopadhyay, A Dutta, EP Jane, SK Biswas. A Profile of 23 Indian Patients with Purpura Fulminans: A Retrospective, Descriptive Study. Indian J Dermatol. 2020; 65: 381-387.
- Nolan J, R Sinclair. Review of management of purpura fulminans and two case reports. Br J Anaesth. 2001; 86: 581-6.
- Chalmers E, Cooper P, Forman K, Grimley C, Khair K, Minford A, et al. Purpura fulminans: recognition, diagnosis and management. Archives of Disease in Childhood. 2011; 96: 1066-1071.
- Price VE, DL Ledingham, A Krümpel, AK Chan. Diagnosis and management of neonatal purpura fulminans. Semin Fetal Neonatal Med. 2011; 16: 318-22.
- Herrera R, PC Hobar, CM Ginsburg. Surgical intervention for the complications of meningococcal-induced purpura fulminans. Pediatr Infect Dis J. 1994; 13: 734-7.
- Rubin LG, W Schaffner. Clinical practice. Care of the asplenic patient. N Engl J Med. 2014; 371: 349-56.
- Gaston MH, Verter JI, Woods G, Pegelow C, Kelleher J, Presbury G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med. 1986; 314: 1593-9.
- Working Party of the British Committee for Standards in Haematology Clinical Haematology Task Force. Guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Bmj. 1996; 312: 430-4.
- Okamura I, Nakamura Y, Katsurada Y, Sato K, Ikeda T, Kimura F, et al. Successful Corticosteroid Treatment for Purpura Fulminans Associated with Quinolone. Intern Med. 2016; 55: 3047-3051.
- Ghosh M, Ray AS, Song M, Varney J, Ghosh S, Koirala J, et al. Staphylococcus-Associated Purpura Fulminans: Can We Expect a Better Outcome With Intravenous Immunoglobulin?. Chest. 2016; 149: A137.
- Werdan K, Pilz G, Bujdoso O, Fraunberger P, Neeser G, Schmieder RE, et al. Score-based immunoglobulin G therapy of patients with sepsis: the SBITS study. Crit Care Med. 2007; 35: 2693-2701.
- Arumugham VB RA. Intravenous Immunoglobulin (IVIG), StatPearls Treasure Island (FL): StatPearls Publishing. 2023.
- Colling ME, PK Bendapudi. Purpura Fulminans: Mechanism and Management of Dysregulated Hemostasis. Transfus Med Rev. 2018; 32: 69-76.
- https://pubmed.ncbi.nlm.nih.gov/1744852/
- Meyer MT, JE Irazuzta, H Tozibikian. Topical nitroglycerin and pain in purpura fulminans. J Pediatr. 1999; 134: 639-41.
- Irazuzta J, ML McManus. Use of topically applied nitroglycerin in the treatment of purpura fulminans. J Pediatr. 1990; 117: 993-5.
- Smith OP, B White. Infectious purpura fulminans: diagnosis and treatment. Br J Haematol. 1999; 104: 202-7.
- Fourrier F, Lestavel P, Chopin C, Marey A, Goudemand J, Rime A, et al. Meningococcemia and purpura fulminans in adults: acute deficiencies of proteins C and S and early treatment with antithrombin III concentrates. Intensive Care Med. 1990; 16: 121-4.
- Cobcroft R, A Henderson, C Solano, D Scott. Meningococcal purpura fulminans treated with antithrombin III concentrate: what is the optimal replacement therapy? Aust NZJ Med. 1994; 24: 575-6.
- Brandtzaeg P, GB Joø, B Brusletto, P Kierulf. Plasminogen activator inhibitor 1 and 2, alpha-2-antiplasmin, plasminogen, and endotoxin levels in systemic meningococcal disease. Thromb Res. 1990; 57: 271-8.
- Aiuto LT, SR Barone, PS Cohen, RA Boxer. Recombinant tissue plasminogen activator restores perfusion in meningococcal purpura fulminans. Crit Care Med. 1997; 25: 1079-82.
- Bernard GR, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001; 344: 699-709.
- Ranieri VM, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012; 366: 2055-64.
- Martí-Carvajal AJ, I Solà, C Gluud, D Lathyris, AF Cardona. Human recombinant protein C for severe sepsis and septic shock in adult and paediatric patients. Cochrane Database Syst Rev. 2012; 2012: Cd004388.
- Smith OP, White B, Vaughan D, Rafferty M, Claffey L, Lyons B, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet. 1997; 350: 1590-3.
- Veldman A, Fischer D, Wong FY, Kreuz W, Sasse M, Eberspacher B, et al. Human protein C concentrate in the treatment of purpura fulminans: a retrospective analysis of safety and outcome in 94 pediatric patients. Crit Care. 2010; 14: R156.
- Chiafery MC, RA Stephany. KJ Holliday. Epidural sympathetic blockade to relieve vascular insufficiency in an infant with purpura fulminans. Crit Care Nurse. 1993; 13: 71-6.
- Anderson CT, CB Berde, NF Sethna, JJ Pribaz. Meningococcal purpura fulminans: treatment of vascular insufficiency in a 2-yr-old child with lumbar epidural sympathetic blockade. Anesthesiology. 1989; 71: 463-4.
- de Chalain T, SR Cohen, FD Burstein. Successful use of leeches in the treatment of purpura fulminans. Ann Plast Surg. 1995; 35: 300-4; discussion 304-6.