
Case Report
Austin J Clin Case Rep. 2024; 11(5): 1332.
Langerhans Cell Histiocytosis in Cervical Vertebra 4 in An Obese Man
Yaya Zhou¹; Zongbo Zhang²; Wendong Xie²; Chao Zhang¹*
¹Department of Orthopedics, Gansu Provincial Hospital, PR China
²Gansu University of Chinese Medicine, PR China
*Corresponding author: Chao Zhang, Department of Orthopedics, Gansu Provincial Hospital, No.204, Donggang WestRoad, Lanzhou, 730000, Gansu Province, PR China. Email: zc-315@163.com
Received: July 22, 2024 Accepted: August 06, 2024 Published: August 13, 2024
Abstract
Langerhans Cell Histiocytosis (LCH) is an uncommon affliction of the immune system, predominantly defined by the atypical proliferation of Langerhans cells various parts of the body. Langerhans cells, integral to the immune system, are typically tasked with the surveillance and elimination of pathogens. In the context of LCH, however, these cells deviate from their normative growth patterns, leading to their unregulated expansion. The etiology of LCH remains obscure, with possible links to genetic predispositions, immunodeficiency, and viral incursions. The clinical manifestations of LCH are protean, primarily presenting as: lymphadenopathy, with enlarged nodes in cervical, axillary, inguinal, and other regions; skeletal involvement, characterized by pain, swelling, and fractures; cutaneous manifestations, such as erythematous papules, plaques, and ulcerations; pulmonary issues, including cough, shortness of breath, and a decline in pulmonary function; and central nervous system impairments, manifesting as headaches, vomiting, vision loss, and paralysis. It is noted that the annual global incidence rate stands at 1 in 200,000 individuals, with the epidemiological data in China yet to be precisely delineated.
Keywords: Langerhans’ cell histiocytosis; Bone destruction; Genetic mutation; Surgical
Introduction
Langerhans' Cell Histiocytosis (LCH) is an array of conditions distinguished by the uncontrolled proliferation, infiltration, and granuloma formation of Langerhans cells, which ultimately results in impaired organ function. The skeletal system (particularly the skull and axial bones), the lungs, the central nervous system (especially the hypothalamic region) and the skin, are among the organs frequently impacted by LCH [1]. LCH can be classified into two principal categories based on the extent of organ involvement: solitary organ disease and multisystemic disease [2]. The former is typically associated with involvement of a single organ, such as the lungs, bones, or skin, and is predominantly observed in adults, often with a more favorable prognosis; conversely, the latter involves multiple systems and is predominantly encountered in pediatric populations [3], portending a less favorable prognosis.
Case Report
The patient was a 32-year-old male with obesity who had been hospitalized for a duration of 16 days due to complaints of neck pain and restricted mobility. He reported the onset of neck discomfort five years prior without a discernible precipitant, with temporary relief achieved through oral analgesics. Imaging studies, including a Computed Tomography (CT) scan and three-dimensional reconstruction, revealed bone destruction at the fourth cervical vertebra, suggestive of a space-occupying lesion (Figure 1a-d). Magnetic Resonance Imaging (MRI) corroborated the presence of abnormal signals at the same cervical level, indicating bone destruction (Figure 1e-i). Upon admission, the patient underwent an examination and resection of the lesion at the fourth cervical vertebra under general anesthesia. Intraoperative findings included severe bone destruction with void formation at the fourth cervical vertebra, without encroachment on adjacent structures (Figure 2a-b). The necrotic bone was meticulously excised using an ultrasonic osteotome, followed by the removal of residual necrotic tissue with an orthopedic curette. The surgical site was then thoroughly irrigated with saline and hydrogen peroxide solution. Subsequently, a titanium cage was implanted into the defect of the fourth cervical vertebra and secured with a cervical locking plate to ensure the stability of the cervical spine. A histopathological examination post-surgery yielded a diagnosis of Langerhans cell histiocytosis (Figure 1H-M). Immunohistochemical staining results were as follows: CD1a (positive), CD138 (negative), D163 (equivocal), CD38 (negative), Langerin (positive), S-100 (positive), Kappa Light Chain (negative), Ki67 (hot spot: 40%), Lambda Light Chain (negative), CD68 (positive), all of which are consistent with Langerhans cell histiocytosis (Figure 3a-i). Molecular testing for BRAF gene mutations identified the V600E mutation type (Figure 3j). Postoperative X-rays taken three months after the procedure demonstrated proper positioning of the hardware with no evidence of new bone destruction (Figure 4).
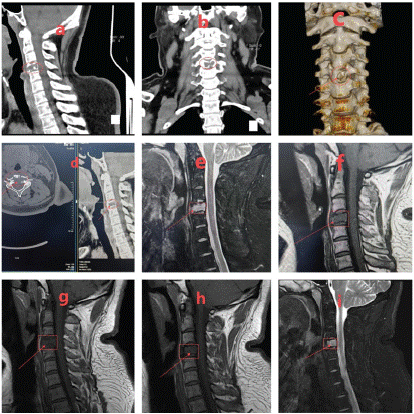
Figure 1: a-d) CT scan and three-dimensional reconstruction revealed evidence of bone destruction of cervical 4 pyramidal, prompting the consideration of a space-occupying lesion; e-i) An MRI scan demonstrated abnormal signal in cervical 4 pyramidal with bone destruction.
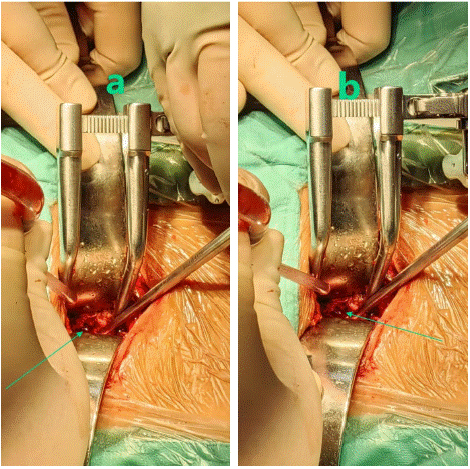
Figure 2: a-b) Visible intra-op: Severe bone destruction of the neck 4 taper with void formation but no violation of the adjacent taper.
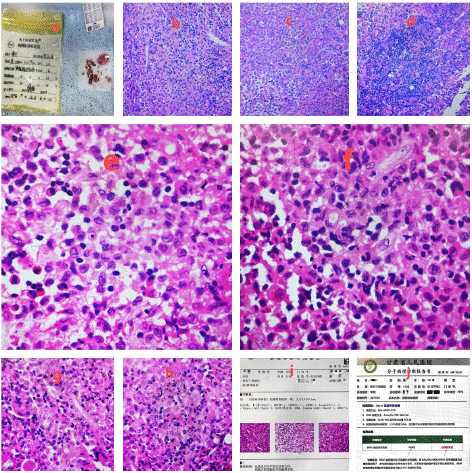
Figure 3: a-i) pathological examination indicated Langerhans cell histiocytosis (Figure 1H-M); immunohistochemical staining indicated [-3] CD1a (+), CD138 (-), D163 (+/-), CD38 (-), Langerin (+), S-100 (+), Kappa Light Chain (-), Ki67 (hot spot: 40%), Lambda Light Chain (-), CD68 (+), consistent with Langerhans cell histiocytosis; j: BRAF gene mutation detection: mutation type was V600E.
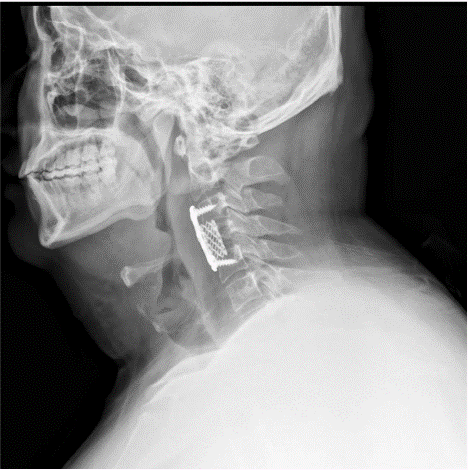
Figure 4: Three months post-op x-rays showed: the hardware was in an optimal position with no evidence of new bone destruction.
Discussion
Diagnosis: The diagnosis of Langerhans Cell Histiocytosis (LCH) is primarily anchored in clinical manifestations, physical findings, imaging studies, and pathological assessment. The cornerstone of diagnosis lies in pathological examination, where the identification of Langerhans cells is facilitated by special staining techniques and immunohistochemical methods.
Treatment: Treatment options for Langerhans cell histiocytosis include pharmacological therapy, radiation therapy, and surgical treatment. The drug therapy mainly includes hormone therapy, chemotherapy and immunosuppressant therapy. The radiotherapy may relieve symptoms for localized lesions. For bone lesions, surgical treatment removes diseased tissue to alleviate pain and risk of fracture.
In this case, it is found that Langerhans cell histiocytosis should be treated actively by surgery when organs or tissues are destroyed, and the effect of drug therapy or radiotherapy is more favorable. Postoperatively, attention should be paid to:
1. Diet: Patients should keep a balanced diet and eat more foods rich in protein, vitamins and minerals to enhance immunity.
2. Rest: Ensure adequate rest and avoid overwork.
3. Prevention of infection: strengthen personal hygiene and avoid contact with viruses such as cold and pneumonia.
4. Regular reexamination: The patient should be examined monitor changes in the condition and to adjust the treatment regimen.
In conclusion, Langerhans cell histiocytosis, as a rare disease, has a small number of patients and is of low concern. However, these patients also need care and support. It requires concerted efforts to raise the society's awareness of rare diseases, care for patients with rare diseases and create a better living environment for them.
Author Statements
Conflict of Interest
The authors declare no financial disclosures or other conflicts of interest relevant to the content of this article to report.
References
- Cohen Aubart F, Idbaih A, Emile JF, Amoura Z, Abdel-Wahab O, Durham BH, et al. Histiocytosis and the nervous system: from diagnosis to targeted therapies. Neuro Oncol. 2021; 23: 1433-1446.
- Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood. 2020; 135: 1319-1331.
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: History, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018; 78: 1035-1044.