
Case Report
Austin J Clin Case Rep. 2017; 4(1): 1114.
Peroneal Muscular Atrophy with Pyramidal Signs: Where Do We Stand
Bhawna Sharma B*, Rahul Jain, Raghavendra BS, Tarun Mathur, Kadam Nagpal and Rahul Handa
Department of Neurology, SMS Medical College Hospital, India
*Corresponding author: Bhawna Sharma B, Department of Neurology, SMS Medical College, Jaipur, India
Received: December 31, 2016; Accepted: February 15, 2017; Published: February 20, 2017
Abstract
Peroneal muscular atrophy with pyramidal features is rare disorder and has been referred to as Hereditary Motor and Sensory Neuropathy (HMSN) type V by Dyck. The HSMN V classification has largely been overshadowed by work in the hereditary spastic paraplegias, specifically those designated “complicated” spastic paraplegia by having both spasticity and other features, including possibly neuropathy. The hereditary neuropathies and spastic paraplegias are groups of genetically diverse disorders classified according to their predominant clinical phenotype and there is considerable overlap between these clinical syndromes in cases where both LMN and UMN features are present in same patient. We report a case of peroneal muscular atrophy with pyramidal features, additionally he had marfanoid features. Association of marfanoid features with HMSN V is not described in literature.
Keywords: Inherited neuropathies; Hereditary sensorimotor neuropathy; Charcoat marie tooth disease; Peroneal muscular atrophy; Hereditary spastic paraparesis
Introduction
Inherited neuropathies are a clinically and genetically heterogeneous group of disorders. The identification of more than 30 causative genes for the inherited neuropathies has raised important questions regarding the approach to their diagnosis [1]. Inherited neuropathies can be broadly classified into two groups: those in which neuropathy is the sole or primary part of the disease (e.g., Charcoat- Marie-Tooth disease, CMT) and those in which the neuropathy is a part of more generalised neurological or multisystem disorder (e.g. familial amyloidotic neuropathy) [1].
Historically, CMT was more commonly called Hereditary Motor and Sensory Neuropathy (HMSN). Dyck and Lambert classify hereditary motor and sensory neuropathies in to seven subgroups in 1968 on the basis of clinical, electrophysiological, pathological and genetic characteristics [2].
Peroneal muscular atrophy with pyramidal features are rare, but should be identified as a distinct disorder [3]. This disorder has been referred to as hereditary motor and sensory neuropathy (HMSN) type V by Dyck [2].
Since the original description of peroneal muscular atrophy by Charcoat, Marie, and Tooth in 1886, it has been well recognised that occasional patients with the syndrome have pyramidal features. This is rare and occurs in less than 5% of cases [4,5].
Current classifications combine the traditional neurophysiological classification with more recently identified genes.
The HSMN V classification has largely been overshadowed by work in the hereditary spastic paraplegias, specifically those designated “complicated” spastic paraplegia by having both spasticity and other features, including possibly neuropathy [2]. Nevertheless, the HMSN V classification may be appropriate in individuals in whom the peroneal atrophy and lower motor neuron involvement is predominant over spasticity [2]. It is relatively benign condition which does not appear to shorten life expectancy or lead to severe disability [3].
Case Presentation
An 18- year old male, born of non consanguineous marriage and uneventful antenatal period presented with delay in motor milestones noticed since the age of 6 months and difficulty in sitting in squatting position and difficulty in walking since early childhood. By the age of 10 year he had stiff gait with tendency to trip. His parents noticed symmetrical wasting of all extremities especially distal, more in lower limbs at around age of 10 year. His symptoms progressed very slowly and he was ambulatory at the age of 18 year and able to do Activity of Daily Living (ADL) independently. He had mild mental retardation. His siblings and other family members were reportedly normal.
General physical examination revealed marfanoid habitus (pectus carniatum, pes planus, and genuvarus), contracture of Achilles tendon and tight hamstrings. No hypertrophy of nerves was observed. All cranial nerves including fundus and visual acuity were normal.
He had symmetrical diffuse atrophy below mid thigh and mid forearm. He had spasticity in all extremities at proximal joints but decreased tone at distal joints. He had symmetrical weakness of all extremities distal more than proximal without any weakness of neck and trunk muscles. Superficial reflexes were present with bilateral extensor plantar response. His supinator and ankle reflexes were absent with brisk biceps, triceps and knee reflexes. His gait was spastic. He had no sensory, cerebellar or extrapyramidal deficit.
His routine haematological and biochemical investigations were normal. Nerve conduction studies revealed significantly delayed distal latencies, decreased CMAPs and uniform slowing of conduction velocities with non recordable F waves in all tested nerves suggestive of demyelinating and axonal affection of lower limbs more than upper limbs. SNAPs were non recordable in all tested nerves on routine NCS. VEP study was normal. MRI brain was normal. MRI spine showed diffuse atrophy of thoracic spinal cord.
Nerve biopsy sections showed disorganised nerve bundles with endoneurialedema and fibrosis. No significant inflammation or granuloma or vasculitis seen (Figure 1 and 2).
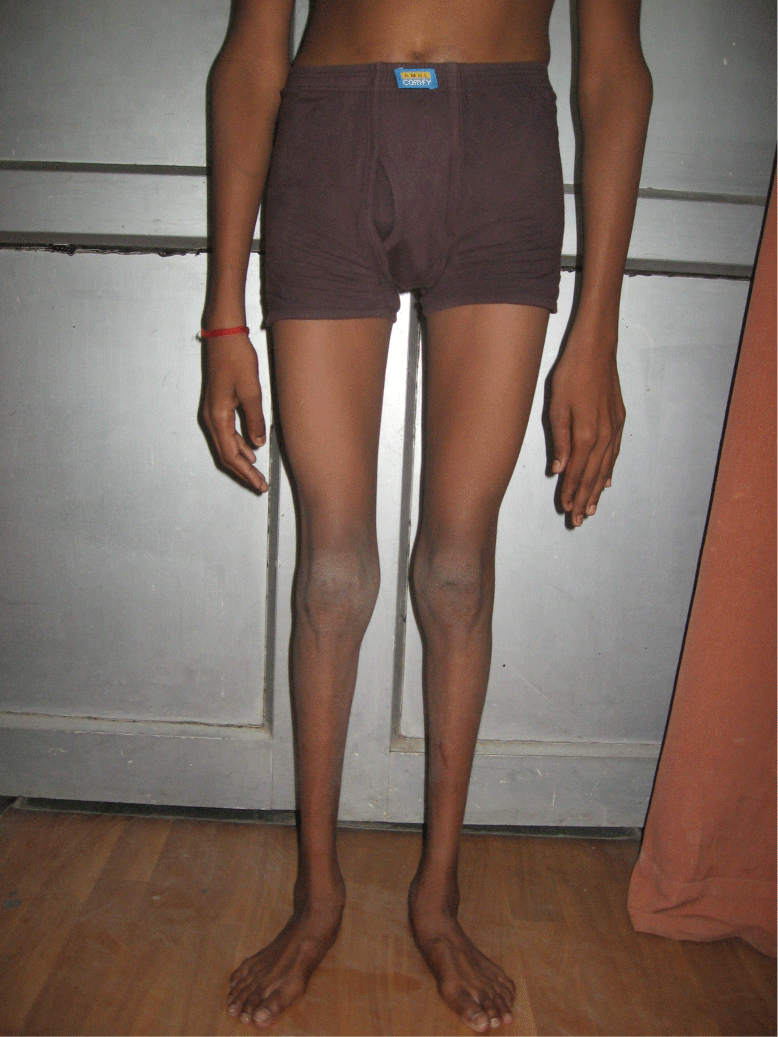
Figure 1: Photograph of patient showing symmetrical distal wasting of
extremities.
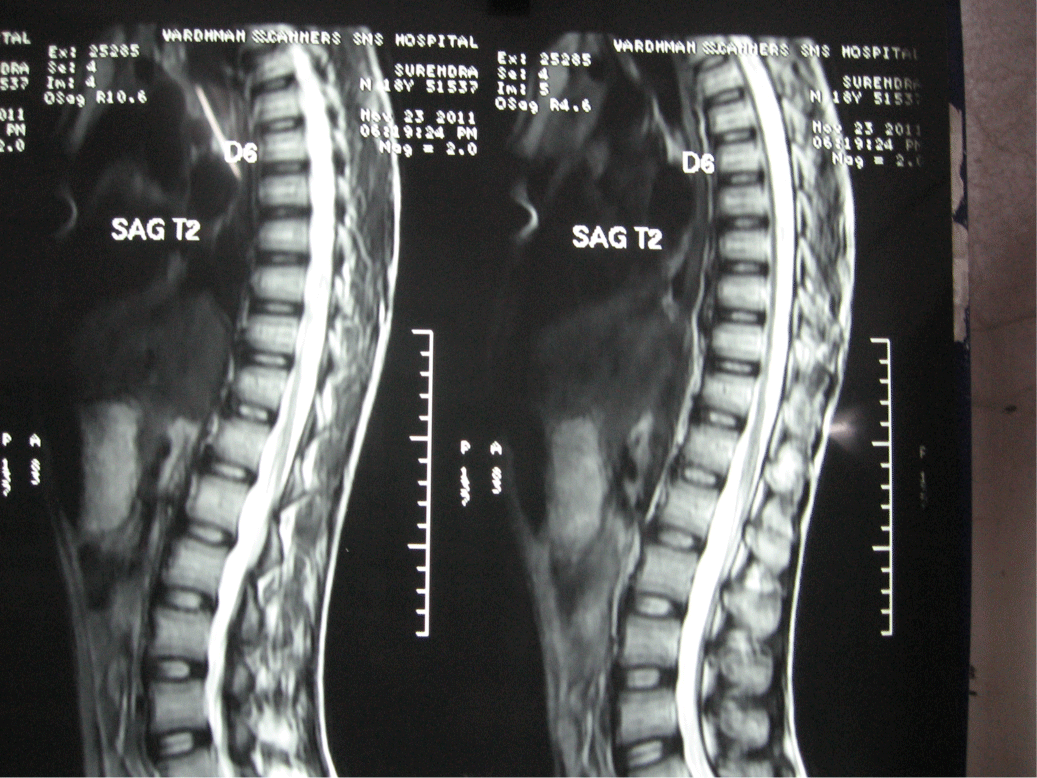
Figure 2: MRI dorsal spine T2 weighted image showing spinal cord atrophy.
Discussion
The patient reported here superficially resemble those with HMSN types I and II (peroneal muscular atrophy). The range of age of onset and distribution of muscle atrophy and weakness are very similar. However another important finding in this patient is presence of significant UMN features in form of brisk reflexes, extensor plantar response and spastic gait. In terms of classification this case of peroneal muscular atrophy with pyramidal features can be allocated to HMSN V, or a case can also be made for classifying this patient amongst the complicated forms of HSP.
Peroneal muscular atrophy with pyramidal features is rare disorders. This disorder has been referred to as hereditary motor and sensory neuropathy (HMSN) type V by Dyck [2]. The HSMN V classification has largely been overshadowed by work in the hereditary spastic paraplegias, specifically those designated “complicated” spastic paraplegia by having both spasticity and other features, including possibly neuropathy [2]. Nevertheless, the HMSN V classification may be appropriate in individuals in whom the peroneal atrophy and lower motor neuron involvement is predominant over spasticity [2]. Our patient had both LMN and UMN features, with LMN features dominating distally over extremities.
The original descriptions of HMSN V noted second decade onset, with autosomal dominant inheritance [2]. The course is typically insidiously progressive over many years. Although walking may be difficult owing to spasticity, many persons are never confined to wheelchair, some become wheel chair bound in the fourth decade or later [2]. Our patient had onset in early childhood and very insidiously progressed, and was ambulatory at the age of 18 years. Our patient did not have any family member affected with similar illness.
Hereditary spastic paraplegias are more commonly designated “uncomplicated” by not having neuropathy or syndromic multiple neurologic system or systemic illness compared to their autosomal recessive and X linked counterparts, in which these features are more common [2].
Pathologic descriptions and Electrophysiologic studies of nerve suggested an axonal peripheral nerve process in cases designated as HMSN V or complicated HSP [2]. Gemignani et al. reported that the entire spectrum of pathologic changes (Hypertrophic type, neuronal type, and Spinal type) in four cases of peroneal muscular atrophy with hereditary spastic paraparesis [6]. In our case nerve biopsy finding was consistent with axonal pathology, but nerve conduction studies revealed findings consistent with both axonal and demyelinating affection.
Other disorders which can have features of both axonal and demyelinating affection are arylsulfatase deficiency (metachromatic leukodystrophy). In our patient, in view of very slow progression, long survival after onset, and lack of white matter changes on MRI brain possibility of metachromatic leukodystrophy was unlikely.
To conclude in our case there were multiple interesting features were noted. Our patient had spastic quadriparesis with bilateral symmetrical wasting distally which fits into HMSN type V and this association is seen in only 5% of all cases of hereditary sensory motor neuropathy. Notably our patient had marfanoid features. To the best of our knowledge any association of marfanoid feature with HMSN V or complicated HSP has not been found. Our patient can also be classified under “complicated HSP” in view of having spastic paraplegia with amyotrophy with evidence of peripheral neuropathy by NCS.
The hereditary neuropathies and spastic paraplegias are groups of genetically diverse disorders classified according to their predominant clinical phenotype. This case further extends the considerable overlap between these clinical syndromes.
References
- Reilly MM. Classification and diagnosis of the inherited neuropathies. Ann Indian Acad Neurol. 2009; 12: 80-88.
- Klein CJ, Dyck PJ. HMSN II (CMT 2) and miscellaneous inherited system atrophies of nerve axon: clinical-molecular genetic correlates. In: Peter J Dyck, P K Thomas, editors. Peripheral neuropathy. 4th edition. Volume 2: Elsevier Saunders; 2004. pp 1742-1743.
- Harding AE, Thomas PK. Peroneal muscular atrophy with pyramidal features. J Neurol Neurosurg Psychiatry. 1984; 47: 168-172.
- Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy type I and II. Brain. 1980; 103: 259-280.
- Davis CJF, Bradley WG, Madrid R. The peroneal muscular atrophy syndrome: clinical, genetic, electrophysiological and nerve biopsy studies. I. Clinical, genetic and electrophysiological findings and classification. J Genet Hum. 1978; 26: 311-349.
- Gemignani F, Guidetti D, Bizzi P, Preda P, Cenacchi G, Marbini A. Peroneal muscular atrophy with hereditary spastic paraparesis (HMSN V) is pathologically heterogeneous. Report of nerve biopsy in four cases and review of the literature. Acta Neuropathol. 1992; 83: 196-201.