
Case Report
Austin J Clin Case Rep. 2019; 6(1): 1140.
Pulmonary Hypertension as a First Presentation for Schistosomiasis: a Case Report of an Unusual Presentation
Mogahed Ismail Hassan Hussein*
Department of Physiology University of Gezira, Division of Internal Medicine, Wad Medani Hospital, Sudan
*Corresponding author: Mogahed Ismail Hassan Hussein, Department of Physiology University of Gezira, Division of Internal Medicine, Wad Medani Hospital, Sudan
Received: January 21, 2019; Accepted: February 20, 2019; Published: February 27, 2019
Abstract
Introduction: The combination of schistosomiasis and pulmonary hypertension was always recognized as a rare one, to the point that studies used to consider pulmonary hypertension as a manifestation of hepatosplenic schistosomiasis but none of them has considered it as a manifestation of Schistosoma infection until recently. In a case series study, 18.5% of patients that have a documented hepatosplenic schistosomiasis were found with pulmonary hypertension. Schistosomiasis rarely causes pulmonary hypertension without evident hepatosplenic manifestations. Here we are reporting a case of a patient whose first clinical presentation was features of pulmonary hypertension. The patient developed SOB and subsequently went on to develop clinical manifestations of the disease, paradoxically without ascites, splenomegaly or any features of the hepatosplenic disease.
Discussion: We use this case as an opportunity to outline pathological mechanisms, causes, and classification of pulmonary hypertension. We also review the literature regarding pulmonary hypertension as a first presentation for schistosomiasis to establish the rarity of this case.
Conclusion: A structured and thorough workup for pulmonary hypertension is emphasized. It is important to exclude all other secondary causes to be able to diagnose primary pulmonary hypertension especially in the absence of a positive family history and advanced diagnostic technology.
Keywords: Schistosomiasis; Pulmonary hypertension; Periportal fibrosis
Abbreviations
APAH: Associated Pulmonary Arterial Hypertension; ECG: Electrocardiogram; FPAH: Familial Pulmonary Arterial Hypertension; IPAH: Idiopathic Pulmonary Arterial Hypertension; LPSH: Left Parasternal Heave; NYHA: New York Heart Association; PAH: Pulmonary Arterial Hypertension; PAP: Pulmonary Arterial Pressure; PMH: Past Medical History; PVR: Pulmonary Vascular Resistance; RV: Right Ventricle; SMCs: Smooth Muscle Cells; SOB: Shortness of Breath; WHO: World Health Organization
Introduction
The blood is pumped from the right ventricle through the lungs and then to the left atrium, which leads to the systemic circulation, this circuit constitute the pulmonary circulation.
The blood from the right ventricle flow through the main pulmonary artery, which then divides into the two pulmonary arteries, one of the supplies the right lung and the other supplies the left. In the lungs, those arteries branch excessively and are connected to arterioles, they lead to capillaries that unite into venules and then veins. Four pulmonary veins, empty the blood from the lungs into the left atrium [1].
As person breathes, the blood in the lung capillaries picks up oxygen. This is why the blood leaving the lung through pulmonary veins have a high oxygen content. In the peripheral circulation, the oxygen is extracted by the tissues, this will decrease the oxygen content of the peripheral veins, the right side of the heart and the pulmonary artery [1].
The resistance is low in the pulmonary circulation owing to the highly distensible vessels. The pulmonary artery has shorter course and thinner walls when we compare it to the aorta, it also contains less smooth muscle and elastin. Pulmonary arterioles also have thin walls, they are not capable of vasoconstriction like systemic arterioles. The pulmonary venules and veins are also very thin and possess little smooth muscle [2].
Pulmonary Circulation in resting healthy individuals, appears to have systolic and diastolic pressure in the pulmonary artery about 25 and 10 mm Hg, respectively, and the mean pressure is about 15 mm Hg. The mean pressure in the left atrium is about 5 mm Hg, and so the total pulmonary arteriovenous pressure gradient is only about 10 mm Hg. The mean hydrostatic pressure in the pulmonary capillaries lies between the pulmonary arterial and pulmonary venous values but somehow it’s closer to the latter [2].
Pulmonary hypertension is considered when the mean pulmonary artery pressure at rest is equal or more than 25 mm Hg [3]. The second WHO conference on pulmonary hypertension, held in Evian, France, in 1998 [4], classified pulmonary hypertension based on similarities in the clinical features [4] and was revised in Venice, Italy, in 2003 to reflect a treatment-based approach to pulmonary hypertension classification [5]. The 4th world symposium on pulmonary hypertension took place in Dana Poi 2008 and provided slight modifications to the classification scheme [6]. This categorization is detailed further in our discussion (Figure 1).

Figure 1: Dana Poi 2008, classification.
Literature Review
In this report, we describe the rare case of a patient who was presented with pulmonary hypertension secondary to Schistosomal infection without any identifiable symptoms or signs of hepatosplenic disease.
A search of PubMed, conducted by the author using the words ‘pulmonary hypertension’ AND ‘schistosomiasis’, restricted to the English Language and to be present in title or abstract resulted in 190 results, 165 excluded based on the title or the abstract, 25 abstracts were reviewed, 20 abstracts were addressing the pathophysiological mechanism behind Schistosomal pulmonary arterial hypertension, three abstracts were focusing on epidemiology of Schistosomal pulmonary arterial hypertension, only two abstracts addressed the timing of the disease presentation, one was titled Early Detection of Schistosoma Egg-Induced Pulmonary Granulomas in a Returning Traveler, and the most interesting article written by Peter P. Turner, concluded that it is not necessary for cirrhosis to be present to allow the ova of S. mansoni to reach the pulmonary arterioles.
Case Presentation
A 26-year-old farmer was presented to the internal medicine department with the complaint of shortness of breath NYHA class IV. He denied any chest pain, palpitations, stridor, syncope, cough or haemoptysis. The patient denies weight loss or systemic features like fevers or malaise. The patient denies abdominal distension, hematemesis, skin changes and dysphagia. He is sexually active with his wife, no PMH of syphilis.
Clinical examination of the cardiovascular and palpation of the precordium shows a palpable P2 and positive LPSH. ECG performed in clinic showed a significant Right axis deviation. Abdominal and hand examinations were normal. Given these clinical findings, it was felt that a pulmonary hypertension was the most likely cause of his symptoms.
ECHO was thereafter performed to estimate the pulmonary artery systolic pressure, which was 28 mm Hg, no RV Dysfunction or any finding that would suggest a cardiac cause.
The patient was concerned about his problem, he received sildenafil tabs and his condition improved. He was also given warfarin as several studies, using both univariate and multivariate analyses, have shown that survival in IPAH, regardless of histopathologic subtype, is increased when patients are treated with anticoagulant therapy [7].
Upon further inquiry at the follow up, the patient revealed that he used to swim and bathe in a local pool. It was concluded that the patient may be an asymptomatic carrier of schistosomiasis, abdominal ultrasound was done and showed normal spleen and liver along with thin periportal fibrosis.
Discussion
The pathological mechanisms of pulmonary hypertension
Although the exact pathogenesis is not yet concluded, many different mechanisms are implicated in the pathogenesis of IPAH, a combination of pulmonary vasoconstriction, in situ thrombosis and pulmonary arterial wall remodelling, which are responsible for the rise in PVR and PAP in patients with PAH, causing a progressive functional decline in patients despite advancing therapies [8].
Pulmonary vascular remodeling: This takes place in the small to mid-sized pulmonary arterioles (‹500 μm). Lesions of the pulmonary vessels occur at the distal and medial pre-capillary arteries, which undergoes various degrees of abnormal muscularisation to the point that some of them are lost. Parts of the pulmonary artery, particularly the lamina of the wall is thickened either in a concentric or eccentric laminar lesion, then the “plexiform lesions” is formed by subsequent fibrinoid necrosis [9]. These changes affect all three layers of the vessel wall (intima, media, and adventitia) as a consequence of cellular hypertrophy, hyperplasia, inflammation, altered energy metabolism, defects in cell differentiation and programmed cell death, migration of cells, and fibrosis. Mutations in the MPR2 gene is the most predisposing factors to develop FPAH and sometimes even the sporadic IPAH. cancer like concept: Lately, scientists had came to a wonderful understanding and released a novel cancer like concept for PAH [10,11] (Figure 2), starting with a monoclonal expansion of endothelial cells in the model with IPAH when compared to the lungs of subjects with congenital heart disease [12]. Also, DNA microsatellite has been obtained indicating instability of short sequences within plexiform lesions in idiopathic PAH [13]. Somatic chromosome abnormalities in the lungs of patients with PAH has been detected [14]; abnormal hyperproliferative, apoptosis-resistant phenotype of the pulmonary endothelial cells and SMCs derived from patients with PAH is present in vitro [15]. Nonetheless, crucial differences between PAH pathogenesis and carcinogenesis have existed, but they are beyond the scope of this article.Vasoconstriction theory: Imbalance between locally produced vasodilators such as nitric oxide and prostacyclin and vasoconstrictors such as endothelin and thromboxane is a major contributor to the observed increase in the pulmonary vascular resistance, and it’s considered one of the pathobiologic basis of therapy.
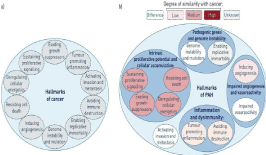
Figure 2: A) In 2000 scientists have proposed a six capabilities that are similar to cancer molecular conditions and they added four additional hallmarks in 2011.
B) Pulmonary arterial hypertension pathophysiology resembles cancer to variable degrees. No invasion or metastasis was observed in PAH. In addition, these
features found in PAH also appear to contribute to the disease pathogenesis with different levels of importance [16].
Pulmonary Arterial Hypertension (PAH)
PAH is a rapidly progressive cardiopulmonary disorder, it has poor prognosis owing to the absence of curative options, the elevated Pulmonary Vascular Resistance (PVR) leads to an elevated Pulmonary Artery Pressure (PAP) leading to cor pulmonale and death [3,12]. The underlying cause maybe pulmonary, cardiac, or systemic disease (associated PAH [APAH], previously known as secondary pulmonary hypertension). On rare occasions, PAH has no identifiable cause or associated underlying disease and here it’s called idiopathic PAH (IPAH) or primary PAH (PPH). A familial form of IPAH (FPAH) accounts for about 6% of cases [17].
The manifestations of the disease are not specific, hence why misdiagnosis may occur. The presence of pulmonary hypertension is clinically suggested by the presence of dyspnea in the absence of heart or respiratory disease. Symptoms can also be fatigue, weakness, angina, syncope, and abdominal distention. Symptoms at rest are reported only in very advanced cases [3]. The physical include left parasternal heave, a loud pulmonary component of the second heart sound (P2), a pan systolic murmur of tricuspid regurgitation, a diastolic murmur of pulmonary insufficiency, and right ventricular S3. Raised JVP, palpable liver, lower limb oedema, ascites causing abdominal distention, and cold extremities are all features of advanced disease. Central cyanosis might also be present. The lung examination is usually normal. The presence of connective tissue diseases, liver cirrhosis, HIV infection, and congenital heart diseases will give a hint to the secondary cause. Abnormal electrocardiographic, chest radiograph or echocardiographic findings may lead to the diagnosis of pulmonary hypertension [3].
Structured algorisms have been released to guide health practitioners to provide more accurate and detailed work up for the disease (Figure 3) [18].
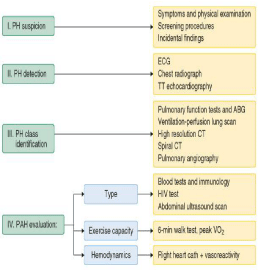
Figure 1: Simplified diagnostic approach to pulmonary hypertension. ABG:
Arterial Blood Gases; Cath: Catheterization; CT: Computed Tomography;
ECG: Electrocardiogram; HIV: Human Immunodeficiency Virus; PH:
Pulmonary Hypertension; PAH: Pulmonary Arterial Hypertension; TT:
Transthoracic; VO2: Oxygen Consumption [19].
Pathophysiology of Schistosomal Pulmonary Arterial Hypertension (PAH)
There are three different pathophysiologic mechanisms behind PH that caused by schistosomiasis. Schistosomal egg migration to pulmonary arteries was the first mechanism to be described. It occurs in case of periportal fibrosis due to parasite-induced granulomas, and it usually not associated with cirrhosis. The egg will cause pulmonary arterial obstruction, which will cause a plexiform lesion [20,21]. Wide and asymmetric vasculopathy is the second proposed mechanism and it was named obliterative arteritis. It may be caused by autoimmune mechanism or associated with egg burden [22]. The final theory proposed that the Schistosomal PH is a variant of porto-pulmonary hypertension. [23]. As the pulmonary arteries have a high flow state, this theory first hypothesised the development of hepatosplenic disease, then portal hypertension and finally pulmonary hypertension, and this explains the cases of PH in the setting of chronic hepatosplenic disease with no evidence of granulomas or egg migration to the pulmonary arteries.
Conclusion
Schistosomiasis is a very common parasitic infection that has a wide range of complications like; systemic anaphylaxis reaction, hepatic, pulmonary, and CNS granulomatous involvement. In endemic areas, the disease may have unusual presentations, like in this case, the patient developed pulmonary hypertension due to schistosomiasis without symptomatic hepatosplenic disease.
From this we can conclude that; in endemic areas, if a patient presents with pulmonary hypertension in the background of exposure to the disease risk factors, the physician must maintain a low threshold for abdominal US to check for periportal fibrosis, and for doing other disease specific investigations.
Funding Statement
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Contributor-ship Statement
D. Hussein has produced the report and literature review. D. Maarib Maki assisted in the literature review and was also involved in the care of the patient.
Disclosures and Ethics
This work has not been published in the form of an abstract or as part of a published lecture or academic thesis or an electronic preprint. It is not under consideration for publication elsewhere, its publication is approved by all authors. It will not be reproduced without the copyright holder permission.
Acknowledgment
We are very thankful to prof. Abdallah Abdalkarem Gebreel at Wad Medani teaching hospital for supervising this work.
References
- Eric P, Widmaier. Physiology, cardiovascular. vander human physiology the mechanism of body function. New York: Mc Graw Hill. 2014.
- Special Circulations. Pulmonary circulation. Achilles J Pappano. Cardiovascular Physiology. Elsevier. 2007.
- Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009.; 54: S55-66.
- Rich S. Executive summary from the World Symposium on Primary Pulmonary Hypertension 1998, Evian, France. World Health Organization. 1998.
- Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004; 43: 5S-12S.
- Simonneau G, Robbins IM, Beghetti M, Channick NR, Delcroix M, Denton PC, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009.
- Byung JK, Yeon-Mok O. Survival benefits of warfarin in Korean patients with idiopathic pulmonary arterial hypertension. Korean J Intern Med. 2015; 30: 837-845.
- Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest, 2012; 122: 4306-4313.
- Guignabert C, Dorfmuller P. Pathology and pathobiology of pulmonary hypertension. Semin Respir Crit Care Med. 2013; 34: 551-559.
- Rai PR, Cool CD, King JA, Stevens T, Burns N, Winn AR, et al. The cancer paradigm of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008; 558-564.
- Sakao S, Tatsumi K. Vascular remodeling in pulmonary arterial hypertension: multiple cancer-like pathways and possible treatment modalities. Int J Cardiol. 2011; 147: 4-12.
- Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NF, Tuder RM. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest. 1998; 927-934.
- Yeager ME, Halley GR, Golpon HA, Voelkel NF, Tuder RM. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res. 2001.
- Aldred MA, Comhair SA, Varella-Garcia M, Asosingh K, Xu W, Noon GP, et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010; 182: 1153-1160.
- Xu W, Koeck T, Lara AR, Neumann D, Difilippo FP, Koo M, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA. 2007; 104: 1342-1347.
- Guignabert C, Tu L, Hiress M, Ricard N, Sattler C, Seferian A, et al. Pathogenesis of pulmonary arterial hypertension: lessons from cancer. Eur Respir Rev. 2013; 22: 1600-1617.
- Ghamra ZW, Dweik RA. Primary pulmonary hypertension: an overview of epidemiology and pathogenesis. Cleve Clin J Med. 2003; 70: S2-S8.
- Galie N, Hoeper MM, Humbert M. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society. Eur Heart J. 2009; 30: 2493-2537.
- Galie N, Torbicki A, Barst R. Guidelines for diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J. 2004; 25: 2243-2278.
- Andrade ZA, Andrade SG. Pathogenesis of schistosomal pulmonary arteritis. Am J Trop Med Hyg. 1970; 9: 305-310.
- Inokuchi T, Maki K, Tsutsumi H. Pulmonary lesions in rabbits with experimental schistosomiasis japonica--derivation of schistosome eggs. Kurume Med J. 1979; 26: 1-9.
- Crosby A, Jones FM, Southwood M, Stewart S, Schermuly R, Butrous G, et al. Pulmonary Vascular Remodeling Correlates with Lung Eggs and Cytokines in Murine Schistosomiasis. Am J Respir Crit Care Med. 2010; 181: 279-288.
- Pereira GA, Bestetti RB, Leite MPB, Santos RB, Ramos SG, Lucchesi FR, et al. Portopulmonary hypertension syndrome in schistosomiasis mansoni. Trans R Soc Trop Med Hyg. 2002; 96: 427-428.