
Case Report
Austin J Clin Case Rep. 2020; 7(1): 1164.
A Patient with Sickle Cell Disease and Beta-Mannosidosis: A Case Report
Almadani H, Namnqani R* and Kurdi H
Department of Pediatrics, King Fahad Armed Forces Hospital, Jeddah, Kingdom of Saudi Arabia
*Corresponding author: Namnqani R, Department of Pediatrics, King Fahad Armed Forces Hospital, Jeddah, Kingdom of Saudi Arabia
Received: April 26, 2020; Accepted: June 23, 2020; Published: June 30, 2020
Abstract
Sickle Cell Disease (SCD) is one of the global health problems, with estimates of approximately 300,000 new cases per year diagnosed with SCD worldwide. It is defined as a homozygous status of the sickle hemoglobin (HbS) gene which results in substitution of the amino acid valine to glutamic acid at the 6th position of the B-globin chain. B-mannosidosis is a rare glycoprotein lysosomal storage disease inherited as an autosomal recessive pattern caused by a deficient activity of beta manosidase enzyme. We report here Eight years old male patient diagnosed with Sickle Cell Disease and Beta-mannosidosis, to our knowledge this is the first case to report a patient with SCD and B-mannosidosis.
Keywords: Sickle Cell Disease; Beta mannosidosis; Hypersplenism; Acute Chest Syndrome; Splenic Sequestration
Introduction
Sickle Cell Disease (SCD) is one of the global health problems, with estimates of approximately 300,000 new cases per year diagnosed with SCD worldwide. It is defined as a homozygous status of the sickle hemoglobin (HbS) gene which results in substitution of the amino acid valine to glutamic acid at the 6th position of the B-globin chain [1,2]. B-mannosidosis is a rare glycoprotein lysosomal storage disease inherited as an autosomal recessive manner caused by a deficient activity of beta manosidase enzyme [3]. Acute Chest Syndrome (ACS) is the leading cause of death in SCD patient and the 2nd most common complication of Sickle Cell Disease, after vasoocclusive crisis [4]. Splenic sequestration and hypersplenism is a known complication as well of SCD [5]. We report a case of SCD with B-mannosidosis who is having a recurrent ACS and hypersplenism with acute splenic sequestration.
Case Presentation
An 8 years old boy, a product of a full term, Spontaneous Vaginal Delivery (SVD), the second child to consanguineous parents. The patient presented at age of 4 months with pallor and shortness of breath with a complete blood count hemoglobin level of 7.8mg/ dl with a sickle cell screen positive, and diagnosed as a sickle cell disease based on hemoglobin electrophoresis (HbS: 34.7% and HbF: 63.7% with no detectable HbA) which was repeated at age of one year and confirmed the diagnosis of SCD with alpha-Thalassemia trait, X-ray chest showed cardiomegaly and Echo was done and results were as dilated left ventricle and atrium with moderate mitral regurgitation and the left ventricle function of FS 17.2% and labelled as having cardiomyopathy. At age of 3 years the patient had global developmental delay, growth parameters were below 3rd centile for height and weight, he had hepatosplenomegaly, speech retardation with hearing loss and recurrent otitis media requiring later bilateral myringotomy, T tube insertion and a hearing aid. He had coarse facial features (gargoyle-like features) as frontal bossing, flat nasal bridge, large tongue with thick lips and gapped teeth, gibbus malformation with skeletal deformities (Figure 1-4) (Table 2).
At 4 years of age he was diagnosed as Bronchial Asthma, Allergic Rhinitis, myopic astigmatism and Attention Deficit Hyperactive Disease (ADHD) with a lower IQ of 70 at age of 7 years. He underwent splenectomy at age of 5 years. The patient was investigated for a lysosomal storage disorder, urine Glycosamino glycan (GAG) was negative for the patient, normal blood Tandem mass spectrometry, and chromosomal analysis of 46, XY. Whole Exome Sequence (WES) detected a homozygous variant of exon 6 of the MANBA gene mutation C.704T>G (p.lle235Arg) and both parents are with a heterozygous status. Enzymatic assay of manosidase activity in serum (Table 1).
![]()
Source
Enzyme
Activity
Reference Range
Serum
Beta-mannosidase
0.004 umol/ml/min
1.49 – 8.33 umol/ml/min
Serum
Alpha-mannosidase
0.14 umol/ml/min
0.1 – 0.2 umol/ml/min
Table 1: Enzymatic assay of Mannosidase enzyme activity in our proband.
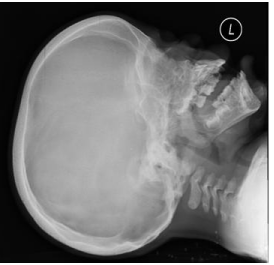
Figure 1: X-ray skull showing calvarial thickening and bullet shaped
vertebrae.
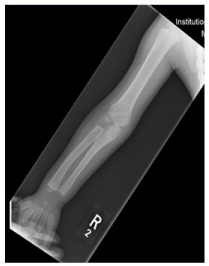
Figure 2: X-ray of right upper limb showing expansion of medullary spaces
with short tubular bones of the hand.
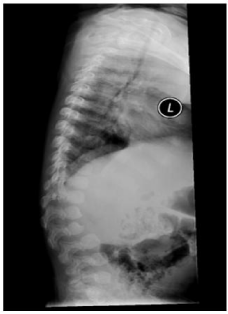
Figure 3: X-ray lateral spine showing L2 vertebral beaking with gibbus
malformation.
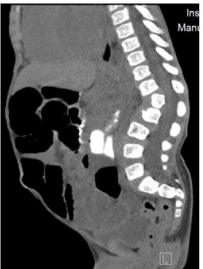
Figure 4: CT Abdomen showing anterior inferior beaking of L2 vertebrae with
gibbus malformation.
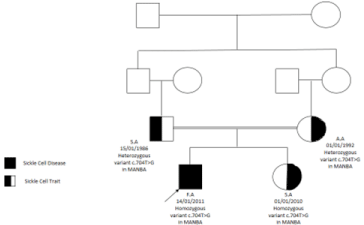
Figure 5: Family pedigree.
He has a sister who had the same presentation of coarse facial features, hepatosplenomegaly, and gibbus malformation with sickle cell Trait and Beta-mannosidosis confirmed by genetic whole exome sequencing (homozygous status of MANBA gene mutation)C. 704T>G) and enzyme assay (Figure 5).
Discussion
Beta-mannosidosis is a pan-ethnic disorder with an autosomal recessive inheritance [6]. Two siblings from Saudi Arabia, described in our report, was associated with gargoyle like facial dysmorphism, hearing impairment, mental retardation, and recurrent respiratory tract infections. To our knowledge the number of patients reported in the literature is small, (Table 2) so, it is difficult to conclude specific symptoms and signs to characterize B-mannosidosis. Various degrees of developmental delay, hearing loss, and mental retardation are common findings. The older sibling has angiokeratoma as an isolated observation as reported previously [21,22]. The observed skeletal changes in our proband are beaked vertebral bodies, thick calvarium, bullet shaped vertebrae, and gibbus malformation (Figure 2).
![]()
Case
1
2
3
4
5
6
7
8
9
10
11
Sex
Male
Male
Male
Male
Male
Female
Male
Female
Female
Male
Male
Ethnicity
European
Hindu
Hindu
Turkish
Turkish
Czech
Czech
Jamaican
Turkish
European
African
Consanguinity
Negative
Positive
positive
Negative
positive
Negative
Negative
Diagnosis age (Yr)
1.5
44
19
8
6
20
30
1
6
3
14
Presenting symptom
Dysmorphology
Mental retardation
Feeding difficulty
Developmental delay
Seizures
Absent speech
Speech impairment
Speech impairment
Mental retardation
Positive
Positive
Yes
Yes
Yes
Positive
Positive
Positive
Positive
Negative
Behavioral problems
Hyperactive
aggressive
Troublesome
Aggressive
Hyperactive aggressive
Hyperactive
Disinterest
Hearing impairment
Mild
Present
Present
Present
Present
Present
Present
Negative
Present
Negative
Negative
Neurological
Speech retard
Speech retard
Developmental delay
Developmental delay
Motor delay
Speech impairment
Peripheral neuropathy
dermatologic
Angiokeratoma
Erysipelas
Facial dysmorphism
Present
negative
Negative
Negative
Negative
Present
Present
Present
Present
Present
Negative
Skeletal deformation
Present
Negative
Negative
Negative
Negative
Present
Present
Negative
Present
Negative
Negative
Respiratory infections
Positive
positive
Positive
Positive
Positive
Positive
Negative
mannosidase
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Urine disaccharides
Positive
Positive
Positive
Positive
positive
Positive
Positive
Positive
Positive
Positive
Faint
Reference
Wenger et al. 1986 [7]
Cooper et al. 1986-1988 [21,22]
Cooper et al. 1986-1988 [21,22]
Dorland et al. 1988 [8]
Dorland et al. 1988 [8]
Kleijer et al. 1990 [6]
Kleijer et al. 1990 [6]
Cooper et al. 1991 [9]
Wijburg et al. 1992 [10]
Poenaru et al. 1992 [11]
Levade et al. 1994 [3]
Table 2A: Beta-mannosidosis phenotype and laboratory findings in reported cases.
![]()
Sibling sister
our case
Case
12
13
14
15
16
17
18
19
20
21
22
23
Sex
Female
Female
Female
Female
Male
Male
Female
Female
Male
Female
female
Male
Ethnicity
white
Arabic
Arabic
Japanese
French
Spanish
Arabic
Algerian
Asian
Arabic
Arabic
Consanguinity
Negative
Positive
Positive
positive
Positive
negative
Positive
Positive
Positive
positive
Positive
Diagnosis age
22 years
7 months
2 years
3.5 years
51 years
18 years
24 years
36 years
12 years
5 months
7 years
5 years
Presenting symptom
clumsiness
Feeding difficulty
Seizures
Seizures
Angiokeratoma
Developmental delay
Angiokeratoma
Angiokeratoma
Clumsiness
Seizures
Dysmorphism
Dysmorphism
Mental retardation
Negative
Positive
positive
Positive
Positive
Negative
Positive
Positive
Positive
Positive
Positive
Behavioral problems
Scantly communicative
ADHD
Negative
Aggressive
ADHD
ADHD
Hearing impairment
Negative
Negative
negative
Present
Present
Mild
Present
Present
Present
Present
Present
Neurological
Negative
Developmental delay
Developmental delay, speech delay
encephalopathy
Peripheral neuropathy
Developmental delay, speech delay
Negative
Cerebellar ataxia
Developmental delay
Developmental delay
Developmental delay
dermatologic
Angiokeratoma
Negative
Negative
Angiokeratoma
Negative
Angiokeratoma
Angiokeratoma
Negative
Angiokeratoma
Negative
Facial dysmorphism
Negative
Negative
Negative
Negative
Negative
Negative
Negative
Negative
Negative
Positive
Positive
Positive
Skeletal deformation
Negative
Negative
Negative
Negative
Negative
Negative
Negative
Negative
Negative
Positive
Positive
Positive
Respiratory infections
Negative
positive
Negative
Negative
Positive
Negative
Positive
Positive
Positive
mannosidase
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Deficient
Urine disaccharide
Positive
Positive
Positive
Positive
Positive
Positive
Reference
Rodriguez et al. 1996 [12]
Gourrier et al. 1997 [13]
Cherian et al. 2003 [14]
Cherian et al. 2003 [14]
Suzuki et al. 2004 [15]
Sedel et al. 2006 [16]
Gort et al. 2006 [17]
Molho et al. 2007 [18]
Levade et al. 2009 [19]
Broomfield et al. 2012 [20]
Almadani et al 2019
Almadani et al. 2019
Table 2B: Beta-mannosidosis phenotype and laboratory findings in reported cases (To our knowledge).
This patient who is a known case of hypersplenism with multiple splenic sequestration with one intensive care admission with critical hemoglobin level mg/dl. Post elective splenectomy the patient had significant decreased rate of admission and blood transfusions. It was noticed that in our proband who has beta-mannosidosis the most frequent complication of sickle cell disease are hypersplenism with sequestration and acute chest syndrome with fewer painful crisis’s although it is the most common feature of sickle cell disease [23]. The patient had frequent admissions as an acute chest syndrome in which after starting azithromycin as an anti-inflammatory agent [24,25] by our pulmonologist, the rate of acute chest syndrome related admission decreased.
The association of Beta mannosidosis and SCD to our knowledge has not been reported before, this combination of both diseases need further reported cases to establish the impact.
Conclusion
This is the first case to report a patient with SCD and B-mannosidosis. We recommend splenectomy for cases of Sickle Cell Disease with B-mannosidosis and hypersplenism, we recommend using azithromycin as an anti-inflammatory prophylactic agent to reduce Acute Chest Syndrome in patient with Sickle Cell Disease and Beta mannosidosis.
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. Consent is available upon request.
References
- Farrell K, Dent L, Nguyen ML, Buchowski M, Bhatt A, Aguinaga MdelP. The relationship of oxygen transport and cardiac index for the prevention of sickle cell crises. J Natl Med Assoc. 2010; 102: 1000-1007.
- Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013; 10: e1001484.
- T. Levade, D. Graber, V. Flurin, M.-B. Delisle, M.-T. Pieraggi, M.-F. Testut, et al. Human b-mannosidase deficiency associated with peripheral neuropathy. Ann. Neurol. 1994; 35: 116–119.
- Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000; 342: 1855-1865.
- Topley JM, Rogers DW, Stevens MC, et al. Acute splenic sequestration and hypersplenism in the first five years in homozygous sickle cell disease. Arch Dis Child. 1981; 56: 765-769.
- WJ Kleijer, P Hu, R Thoomes, M Boer, JGM Huijmans, W Blom, et al. B-Mannosidase deficiency: heterogeneous manifestations in the first female patient and her brother. J. Inherit. Metab. Dis. 1990; 13: 867–872.
- D.A. Wenger, E. Sujansky, P.V. Fennessey, J.N. Thompson, Human b-mannosidase deficiency. N. Engl. J. Med. 1986; 315: 1201–1205.
- L. Dorland, M. Duran, F.E.T. Hoefnagels, J.N. Breg, H. Fabery de Jonge, K. Cransberg, et al. b- Mannosidosis in two brothers with hearing loss. J. Inherit. Metab. Dis. 1988; 11: 255–258.
- Cooper, J.E. Wraith, W.J. Savage, M. Thornley, M.J. Noronha. b-Mannosidase deficiency in a female infant with epileptic encephalopathy. J. Inherit. Metab. Dis. 1991; 14: 18–22.
- H. Wijburg, J. De Jong, R. Wevers, J. Bakkeren, F. Trijbels, R. Sengers. b-Mannosidosis and ethanolaminuria in a female patient. Eur. J. Pediatr. 1992; 151: 311.
- L. Poenaru, S. Akli, F. Rocchiccioli, P. Eydoux, P. Zamet. Human b-mannosidosis: a 3-year-old boy with speech impairment and emotional instability. Clin. Genet. 1992; 41: 331–334.
- M Rodriguez-Serna, R Botella-Estrada, A Chabas, MJ. Coll, V. Oliver, MI. Febrer, et al. Angiokeratoma Corporis Diffusum associated with b-mannosidase deficiency. Arch. Dermatol. 1996; 132: 1219–1222.
- E. Gourrier, M.P. Thomas, A. Munnich, L. Poenaru, D. Asensi, D. Jan, et al. A new case of b mannosidosis. Arch. Pediatr. 1997; 4: 147–151.
- M.P Cherian. β-Mannosidase Deficiency in Two Mentally Retarded Girls with Intractable Seizures. Ann Saudi Med. 2004; 24: 393-395.
- Suzuki, Nuiko & Konohana, Izumi & Fukushige, Tomoko & Kanzaki, Tamotsu. B-Mannosidosis with Angiokeratoma Corporis Diffusum. The Journal of dermatology. 2004; 31: 931-935.
- Sedel F, Friderici K, Nummy K, et al. Atypical Gilles de la Tourette Syndrome With β-Mannosidase Deficiency. Arch Neurol. 2006; 63: 129–131.
- Gort, Laura & Duque, Joana & M Fabeiro, José & Zulaica, Ander & Josep Coll, Maria & Chabás, Amparo. Molecular analysis in two β-mannosidosis patients: Description of a new adult case. Molecular genetics and metabolism. 2007; 89: 398-400.
- Molho, Vered & Bargal, Ruth & Abramowitz, Yigal & Doviner, Victoria & Ingber, Arieh & Raas-Rothschild, et al. Angiokeratoma corporis diffusum in human β-mannosidosis: Report of a new case and a novel mutation. Journal of the American Academy of Dermatology. 2007; 57: 407-412.
- Labauge, Pierre & Renard, Dimitri & Castelnovo, Giovanni & Sabourdy, Frédérique & Menjot de Champfleur, Nicolas & Levade, Thierry. Betamannosidosis: A new cause of spinocerebellar ataxia. Clinical neurology and neurosurgery. 2008; 111: 109-110.
- Broomfield, Alexander & Gunny, R & Ali, I & Vellodi, Ashok & Prabhakar, Prab. A Clinically Severe Variant of β-Mannosidosis, Presenting with Neonatal Onset Epilepsy with Subsequent Evolution of Hydrocephalus. JIMD reports. 2013; 11.
- Cooper, I.B. Sardharwalla, M.M. Roberts. Human b-mannosidase deficiency. N. Engl. J. Med. 1986; 315: 1231.
- Cooper A, Hatton C, Thornley M and Sardharwalla IB. Human fl-mannosidase deficiency: Biochemical findings in plasma, fibroblasts, white cells and urine. J. Inher. Metab. Dis. 1988; 1: 17-29.
- Brousseau DC, Owens PL, Mosso AL, Panepinto JA, Steiner CA. Acute care utilization and rehospitalizations for sickle cell disease. JAMA. 2010; 303: 1288-1294.
- Culic O, Erakovic V, Cepelak I, et al. Azithromycin modulates neutrophil function and circulating inflammatory mediators in healthy human subjects. Eur J Pharmacol. 2002: 450: 277-289.
- Pene Dumitrescu T, et al. Development of a population pharmacokinetic model to describe azithromycin whole-blood and plasma concentrations over time in healthy subjects. Antimicrob. Agents Chemother. 2013; 57: 3194– 3201.