
Case Report
Austin J Clin Case Rep. 2020; 7(3): 1172.
Neck Fusocellular Fibrosarcoma - A Rare Presentation of a Rare Disease
Marta Reia¹*, Carlos Quintana² and Carlos Penalva Santos³
1Department of General Surgery, Extremadura University, Spain
2Department of Anatomical Pathology, Espirito Santo Hospital, Portugal
3Department of General Surgery, Santa Luzia Hospital, Portugal
*Corresponding author: Marta Reia, Department of General Surgery, Santa Luzia Hospital, Elvas, Extremadura University, Spain. Largo Ricardo Vaz Monteiro nº12, 7330-260 Santo Antonio das Areias, Portugal
Received: May 07, 2020; Accepted: October 09, 2020; Published: October 16, 2020
Abstract
Sarcomas are an uncommon diversified group of neoplasms, which affects soft tissue: muscle, fatty tissue, lymphatics, blood vessels and nerves. Soft Tissue Sarcomas (STS) account for approximately 1% of all adults malignancies. They are hard to diagnose, requiring image exams, and imuno-hystochemical and molecular tests to confirm the diagnosis. The treatment is multidisciplinary, involving surgery, radiotherapy, chemotherapy, and monoclonal therapy, depending on the presenting stage of the disease. This article reports a clinical case of a fusocellular fibrosarcoma in a 20-year-old female, presenting with a cervical indolent mass. The histologic exam revealed an incomplete excision of the lesion (R1) and it showed aspects related to an undiferentiated fusocellular fibrossarcoma, (G3).
This case was discussed in multidisciplinary consultation and the patient was referred to a Sarcoma Group in a tertiary center, where a new surgery was performed for excision of the remaining disease. The anatomo-pathologic review showed a complete excision (R0), but with an insuficient margin, so that the patient was reoperated to obtain larger margins. The pathological exam of this new sample was negative for atypia. After surgical treatment, it was decided by the multidisciplinary team adjuvant treatment with radiotherapy. Follow-up: without evidence of disease relapse until today, eight years after diagnosis. This case is relevant regarding the rarity of this tumours and the importance of obtaining sufficient margin to avoid relapse. We may remain unnoticed because of its indolent presentation, which can delay the diagnosis, several times presenting with advanced disease, reducing therapeutic options and survival.
Keywords: Neck soft tissue; Fibrosarcoma; Fusocellular fibrosarcoma; Fusocellular mesenquimous sarcoma
Abbreviations
STS: Soft Tissue Sarcomas; MRI: Magnetic Resonance Imaging; CT scan: Computorized Tomography scan; WHO: World Health Organization; FNCLCC: French Federation Nationale des Centre de Lutte Contre le Cancer; NCI: National Cancer Institute; UICC: Union for International Cancer Control; AJCC: American Joint Committee on Cancer
Introduction
Sarcomas are un uncommon diversified group of neoplasms, which affects soft tissue: muscle, fatty tissue, lymphatics, blood vessels and nerves. The anatomic distribution include the extremities (43%), the trunk (10%), visceral (19%), retroperitoneum (15%) or head and neck (9%) [1]. Soft Tissue Sarcomas (STS) account for approximately 1% of all adults malignancies [1-3]. The true incidence of sarcomas is underestimated, in Europe being estimated in 4,5/1000 per year [4]. The main factor risks are hereditary syndromes like retinoblastoma, neurofibromatosis type I, tuberous sclerosis, familiar adenomatous polyposis, Gardner syndrome, Li Fraumeni syndrome, Wegener syndrome and Gorlin Syndrome. Previous radiotherapy treatments are also an important risk factor. The majority of sarcomas are clinically asymptomatic, but they can present as rapidly growing lesions, compressing nearby organs or spread in the form of secondary lesions. STS have hematogenous spreading pattern and most commonly metastasizes to the lungs [1].
Soft tissue sarcomas are hard to diagnose, requiring image exams, and immuno- hystochemical and molecular tests to confirm the diagnosis. The treatment is multidisciplinary, involving surgery, radiotherapy, chemotherapy, and monoclonal therapy, depending on the stage of the disease.
Case Presentation
This case report: 20-year-old female patient, without previous medical or familiar history, whom was referred to consultation with a painless mass in the posterior cervical region. In the physical exam, this was an indurated mass, but not of petrous consistency, painless at palpation, but adherent to the superficial and deep surrounding tissue. No inflammatory sings were present, neither it was haemorrhagic. As part of complementary study, the patient did an ultrasound which revealed an ovalated lesion, with 61mm in diameter, regular border, hypoechogenic, heterogeneous and with central vascularization. Regarding the apparent benign sings described, the patient underwent surgical excision of this lesion, without complications. The histologic exam revealed an incomplete excision of the lesion (R1) and it showed aspects related to an undiferentiated fusocellular fibrossarcoma, (G3). The detailed report revealed an infiltrative mesenquimatous pattern of proliferation, fusocellular, hypercelular and fasciculated, with moderate nuclear polymorphism; scattered proeminent nucleolus, and fibro-hyalin stroma. There were necrotic areas, and a mitotic index of 30 figures/10 hpf (Figures 1 and 2).

Figure 1: Magnifying glass image: poorly delineated borders, with
multinodular architecture, composed by hypercellular nodes, alternated with
less cellular areas. The blue ink delineates the margin of surgical excision.
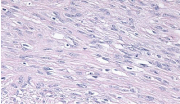
Figure 2: Microscope image: Neoplasic cells with light and eosinophilic
cytoplasm, with poorely defined borders, ablongated and vesiculou nucleous,
with fine cromatin and visible nucleolus.
The immuno-hystochemical study was positive for markers BCL- 2, CD-99 and vimentin; it showed a Ki-67 of 25%. It was negative for markers pancytokeratin AE1/AE3, cytokeratin 7, cytokeratin 20, CAM5.2, S100, CD45, CD20, CD34, CD31, CD117, CD57 CD68, actin, desmin, myoglobin, EMA, FXIII and collagen IV (Figures 3 and 4).

Figure 3: Positive immuno-hystochemical analysis for marker BCL-2.

Figure 4: Positive immuno-hystochemical analysis for marker Vimentin.
This case was then discussed in multidisciplinary consultation in the Hospital of Santa Luzia, Elvas, and it was decided to refer to a tertiary center - Sarcoma Group Consultation, at the Portuguese Institute of Oncology, in Coimbra - for further treatment.
The patient underwent oncologic staging with cervical Magnetic Resonance (MRI) and thoraco-abdomino - pelvic CT scan. The cervical MRI showed subcicatricial residual lesion, 15x7mm, no signs of lymphatic invasion, nor other lesions; and the CT scan also excluded secondary disease. After multidisciplinary evaluation, the decision was to perform new surgery for excision of the remaining disease. The anatomo-pathologic review showed a complete excision, but with an insuficient margin, and the patient was reoperated to obtain larger margins, which went without complications. The pathological exam of this new sample was negative for atypia. After this surgical treatment, it was decided by the multidisciplinary team adjuvante treatment with radiotherapy. Until the present date, eight years after surgery, the patient remains us follow-up at the Sarcoma Group Consultation, with no further incidents.
Discussion
Soft tissue chapter is one of the most complex and demanding of Pathological Anatomy, with multiple entities with distinct biologic behaviour. In 2013, the World Health Organization (WHO) established a new soft tissue tumours classification, stressing the detailed clinical, histological and genetic features of this tumours, made possible by the most recent information provided by cytogenetic and molecular. Additionally, this classification includes a revised categorization of biological behaviour, which allows for two distinct types of intermediate malignancy [2].
Benign mesenchymal tumours outnumber sarcomas by at least 100. Soft tissue sarcomas become more frequent with increasing age, and about 10% of the patients present with detectable metastasis at the time of diagnosis, usually in the lungs [2]. The etiology is unknown, but multiple causes are refered, including genetic and environmental factors, irradiation, viral infections (HPV8, EBV) and immune deficiency. Irrespectively of this causes, the majority of soft tissue sarcomas seem to arise de novo, without an apparent causative factor [2]. Clinically, this lesions commonly present as painless, accidentally detected tumours, without functional defects or repercussion in general health, despite the often large tumour volume. This lesions are better characterized by Magnetic Resonance Imaging (MRI), which has the ability to distinguish tumours tissue from adjacente muscle and fat, besides the information regarding the lesion size, relationship to muscle compartments, fascial planes, and bone and neurovascular structures. It also provides data about the intrinsic characteristics of the tumour (hemorrhage, necrosis, edema, cystic and myxoid degeneration, and fibrosis).
The CT scan is performed at the time of the diagnosis, searching for the evidence of lung metastasis. PET is also a valuable exam, giving information regarding the biological activity of soft tissue masses. The histologic characterization of this masses is of paramount importance, for the accurate diagnosis, tailored treatment, and prognostic implications. This is better obtained with a core biopsy, and fine needle aspiration is not usually performed, given its limited sampling. The pathologic assessment of this samples is often dificult, so that several techniques have been used as an adjuvant to morphologic diagnosis (IHC, cytogenetics, molecular genetics). The new WHO classification redefines the biological potential of this tumours, classifying them as benign, intermediate (locally aggressive), intermediate (rarely malignant), and malignant. Additionally, the histological grading of soft tissue tumours is obtained for its prognostic implications. The FNCLCC (French Federation Nationale des Centre de Lutte Contre le Cancer) and NCI (United States National Cancer Institute) grading systems are the two most widely used systems for this purpose, providing a score and grade after evaluation of the tumour differentiation, mitotic rate and amount of tumour necrosis [1,2]. With all this information, the disease may be staged. The major staging system used is the one developed by the Union for International Cancer Control (UICC) and the American Joint Committee on Cancer (AJCC), clinically useful and of prognostic value, using the TNM system [1,2]. Surgery remains the standard treatment of soft tissue tumours, but radiotherapy and chemotherapy are also often indicated, specially in bulkier and high-grade lesions. This approach is defined by an expert multidisciplinary team. A properly executed surgical resection is the most importante part of the overall treatment, with clear margins from tumour (preferentially >1cm), dictated by the size, anatomical location, relation with normal structures, and functional implications. The need for reconstructive surgical procedures may be considered. The margin status is the most important prognostic factor. In areas where severe loss of function is likely, or the lesion is adjacent to major neurovascular bundles, the role of adjuvant or neoadjuvant radiotherapy and/or chemotherapy becomes more relevant, to downstage larger lesions and reduce the probability of local failure. More recentely, targeted therapy have also shown promising results in patients with certain histologic types of advanced or metastatic soft tissue tumours [1]. Fibrossarcoma represent about 10% of musculoskeletal sarcomas, diagnosed more commonly in patients in the fourth decade. It may present as a soft tissue mass or as a bone tumour. When it affects soft tissue, the age spectrum is wider [5].
As many other sarcomas, soft tissue fibrossarcoma often present as a painless mass, frequently arising deep to the muscular fascia, and may become extremly large until the diagnose is obtained [5].
Fibrossarcoma originates from mesenchymal cells, which transforms in tumours of malignant fibroblasts and collagen. Welldifferentiated forms have multiple plump fibroblastos with deeply staining nuclei in a rich collagen background, but high grade lesions are very cellular, with marked cellular atypia and mitotic activity, with sparse matrix [4]. The fusocellular variant is an uncommon variant within fibrosarcomas, with fish-bone-like cell fascicules. This kind of cell morphology is detected in many other sarcomas (malignant fibrous histiocytoma, leiomyosarcoma, monohpasic fase of synovial sarcoma, neurogenic sarcoma), and differential diagnosis is not always easy [6]. The treatment strategy is the same as stated previously, requiring optimal biopsy and surgery, with appropriate margins. In soft tissue lesions, radiation therapy is often employed as adjuvant treatment, while chemotherapy is used in higher grade lesions [5].
Conclusion
In this case report, the definitive diagnostic was a soft tissue fusocellular fibrossarcoma, stage IA: T1 N0 M0 G3 [FNCLCC score 7: Tumour undifferentiation 3 (high-grade), Mitotic count 3 (regarding >20 mitosis per 10hpf), Tumour necrosis 1 (<50% tumour necrosis)]. The initial approach has been the excision of a primitive lesion of the skin, which revealed the fibrossarcoma, followed by completion of resection requiring additional margin enlargement and adjuvante radiotherapy, with no signs of relapse until today.
Acknowledgement
We would like to show our gratitude to Dr. Carlos Quintana (Head of Department) from Anatomical Pathology Department of the Espírito Santo Hospital in Evora, Portugal, for providing the images of the histopathologic analysis of this case´s specimen.
References
- National Comprehensive Cancer Center (NCCN) Guidelines, Clinical Practice Guidelines in Oncology, Soft Tissue Sarcoma; Version 6.2019; 2020.
- World Health Organization (WHO) classification of soft tissue tumours, 2013.
- Ryan, C and Meyer J. Clinical presentation, histopathology, diagnostic evaluation, and staging of soft tissue sarcoma; Up To Date; 2020.
- Casali P, Abecassis N, Bauer S, Biagini R, Bielack S and Bonvalot S, et al. Soft tissue and visceral sarcomas: ESMO–EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up; Annals of Oncology. 2018; 29: 51–67.
- Dickey I. Fibrosarcoma. E-medicine, Medscape. 2018.
- Ramon Y, Cajal S. Tumores de celulas fusiformes. Servicio de Anatomia Patologica del Hospital Universitario Vall d’Hebron, Barcelona and Sociedad Espanola de Anatomia Patologica; Spain.