
Case Report
Austin J Clin Case Rep. 2020; 7(4): 1178.
A Case of Agyria-Pachygyria Presenting as Seizure Disorder in a Young Girl
Shikha Pandey1, Nitesh Raj Dulal1, Mohan Bhusal1 and PVS Rana2*
1Internal Medicine, College of Medical Sciences, Nepal
2Neurophysician, College of Medical Sciences, Nepal
*Corresponding author: P.V.S. Rana, Department of Neurology, College of Medical Sciences, Nepal
Received: October 19, 2020; Accepted: November 10, 2020; Published: November 17, 2020
Abstract
“Lissencephaly”, a rare gene linked defective neuroblast migration disorder resulting in defective cortical lamination, abnormal gyral development and subcortical heterotropia. Advances in molecular genetics have led to the identification of lissencephaly gene on chromosome 17p 13.3 and causing Type-1 Lissencephaly or miller Diecker syndrome where lissencephaly is severe in posterior brain region. Another X-linked gene Double Cortin (DCX) gene where the lissencephaly is more severe in anterior region of the brain. Usually this defect manifests in early infancy or childhood as seizure disorder. A case of lissencephaly with features of Miller Dieker syndrome in a young girl is reported and literature is reviewed. The important feature of the case was its late presentation in a 17 years old girl.
Keywords: Agyria; Cortical Dysplasia; Seizure; Lissencephaly (LIS); Miller-Dieker Syndrome (MDS); Pachygyria; Polygyria
Introduction
Lissencephaly (LIS), a combination of two Greek words (Lissos = smooth: Encephalin = brain), is a developmental anomaly of brain resulting from abnormalities of neuronal migration [1] and is characterized by absent (agyria) or broad, flat and thick gyri (pachygyria). It is a rare disease with a prevalence rate of 12 per million births [2]. The affected children have varied neurological manifestations depending upon the severity of conditions and associated abnormalities. Rarity of condition led to this case report.
Case Summary
Ms. SC, a 17 years old girl was admitted in Neurology centre of Collage of Medical Sciences (COMS), Bharatpur, (Chitwan-District), Nepal on 16 January 2016 following a seizure episode. As per informant (i.e. her father) her illness started 03 years back when she started having recurrent seizures with a frequency of 3-4 seizures per month. Her old documents were not available but according to history after initial episode she was put on carbamazepine 200 mg twice a day. As her seizures were uncontrolled the dose of carbamazepine was increased in a stepwise manner to 400 mg twice a day. Inspite of increasing dose of carbamazepine her seizures were uncontrolled and were occurring at irregular intervals. When reported she was not taking carbamazepine for the last 3-4 weeks. The seizure semiology suggested complex partial seizure with loss of consciousness. Her seizure started with staring look followed by turning of face to left side, stiffening of the left half of the body and loss of consciousness. She denied history of inter-ictal tongue bite, sphincteric incontinence, post-ictal paralysis or automatism. There was no history of birth trauma or major systemic illness in the past. Family history for similar illness was negative. Her milestones were normal but her scholastic performance was poor and she dropped out after 7th class. Neurological examination revealed facial paresis of upper motor neuron type and hemiparesis (power grade 4/5) on right side. Deep tendon reflexes were brisk on right side with plantar response mute on both sides. Fundus examination was normal. No phakoma detected. Other systems were normal. Investigations done i.e. complete haemogram, urinalysis, metabolic parameters and X-ray chest were normal. EEG (Figure 1) showed dysrhythmia with frequent seizure discharges arising from both sides. No phenomenon of phase reversal, periodic discharges or suppression burst noted. MRI (Figure 2) revealed abnormally thickened gyrus (Pachygyria) involving insular, temporal and parietal region. She was put on sodium valproate (a drug of choice for her seizure type as carbamazepine was ineffective). Her seizures were satisfactorily controlled by sodium valproate 400 mg/twice daily.
Discussion
Lissencephaly is a cortical dysplasia resulting from defective migration of neuronal precursor (neuroblast) from germinal matrix (lining the lateral and 3rd ventricles) along radial glial fibers (extending from ventricles to brain surface) to form cortex between 12 th & 24th week of gestation [1,2]. A completely smooth brain, devoid of sulci and gyri (complete lissencephaly) is rarely seen. Most of the patients have incomplete lissencephaly with agyria and pachygyria in varied combination. Present classification of lissencephaly is based on MRI findings and on associated developmental anomalies [3,4].
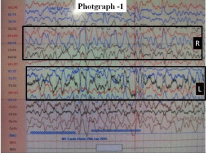
Figure 1: EEG showing markedly disorganized background activity with
seizure activity.
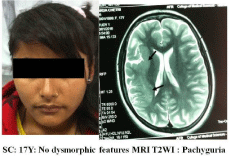
Figure 2: MRI Showing thickened gyrus in right posterior frontal and anterior
parietal region.
Damaska (1983) [3] classified Lissencephaly into Type-1 (classical) lissencephaly having agyria- pachygyria in varied combination with Miller-Dieker syndrome as its prototype and Type-II (Cobblestone) lissencephaly with polymicrogyria (excessive number of abnormal small gyri) . Dietrich et al., (1992) [4] classified lissencephaly into i.e. Type-I lissencephaly showing colpocephaly, thickened cortex with broad flatten gyri, smooth grey white matter inter surface and straight, oblique or shallow sylvian fissure giving a figure of eight appearance; Type-II lisssencephaly with polymicrogyria and Type-III lissencephaly with microcephaly, moderately thick cortex and hypoplasia of cerebellum and brainstem. Additional developmental anomalies reported include Miller-Dieker syndrome* with Type-1, Walker- lissencephaly respectively. For above classifications, pachygyria needs to be distinguished from polymicrogyria which at time is difficult to distinguish by MRI, hence it was suggested to reclassify Type-1 lissencephaly,having only agyria-pachygyria as non lissencephaly cortical dysplasia [5].
Many genetic mutations have been reported in lissencephaly cases [6-8]. Two major genes implicated in classical lissencephaly are LISI on Chromosome 17 and Doublecortin (DBX) or XLIS gene on chromosome X. While larger deletion of LISI gene toward telomere 17p causes Miller-Dieker syndrome, smaller deletion or point mutation of LISI and mutation of XLIS in affected males leads to lissencephaly-17 and lissencephaly-X respectively. These cases have normal facial appearance or may have subtle abnormalities which are different than reported in Miller-Dieker Syndrome. Other genetic abnormalities encountered are (a) mutation of TUBA1A gene with posteriorly dominant gyral pattern [8]; (b) RELIN gene mutation which is associated with autosomal recessive lissencephaly and cerebellar hypoplasia [9] and (c) ARX gene mutations which are associated with X-linked lissencephaly, abnormal genitalia and corpus callosum agenesis.
Children with classical lissencephaly (Miller Dieker Syndrome) have typical dysmorphic features (microcephaly, mid face hypoplasia, small upturned nose, low set small ears, small jaw, thick upper lips and anomalies affecting other organs) and presents with severe mental retardation, motor disabilities varying from severe hypotonic to severe spastic paraplegia, feeding problems and failure to thrive. The case under discussion has MRI findings of classical Lissencephaly but she had no dysmorphic feature Miller-Dieker syndrome.
In Miller-Dieker syndrome seizures occurs in over 90% of cases with onset before 06 months in about 75% of cases [7]. Typically seizures begins in first month of life as massive bilateral myoclonus or infantile spasms in 80% cases but without typical hypsarrhythmia on EEG [7,10]. The EEG is in these patients show characteristic high amplitude fast activity, predominantly in alpha and beta range persisting on eye opening and during sleep phase. The sleep phase lack vortex wave, spindling formation and theta slowing. A 14 CPS activity may be seen. With age the faster activity increases to 25-30 HZ over posterior region with alfa activity is noted in rolandic and parietal region [10].
Though developmental anomaly was present since birth, in present case it presented with multiple seizure types when patient was 13 years old. Similar multiple type seizures are recorded in older children. The EEG showed disorganized background activity consisting of high voltage activity of 5-6 HZ and seizure activity. Earlier these cases remained undiagnosed and such patients were considered having seizure disorder of idiopathic etiology.
References
- Barkovich AJ, Greesens P, Evrard P. Formation, maturation and disorders of brain neocortex. AJNR. 1992; 13: 423-446.
- Guerrini R, Parrini E. Neurogenetics of Epilepsy In Ed Kennard C “ Oxford Text Book of Epilepsy and Epileptic seizures Oxford University Press. 2013 (Reprinted 2014): 11-26.
- Damaska M, Wisniewski K, Sher J. Lissencephaly: Two distinct clinicopathological types. Brain Dev (Tokyo). 1983; 5: 302-310.
- Dietrich RB, Demos D, Kocit et al. Lissencephaly: MRI and CT appearance with different subtypes. Radiol. 1992; 185: 123-126. Quoted from Disorders of diverticulum and cleavage, sulcation and cellular migration Eds Osborn AG, Diagnostic Radiology, AG, Mosby (Elsevier), 3rd Edition. 2008; 3: 37-58.
- Barkovich AJ, Kjos, BO. Non lissencephalic cortical dysplasia’s: correlation of imaging findings with neurological deficits. AJNR. 1992: 13: 95-103.
- Forman MS, Squier W, Dobyns WB, Golden JA. Genotypically defined lissencephalies show distinct pathologies. J. Neuropathol. Exp. Neurol. 2005; 64: 847-857.
- Guerrini R, Fillipi T. Neuronal migration disorders, genetics and elileptogenesis. J Child Neurol. 2005; 364: 287-299.
- Poirier K, Keays DA. Francis-Saildour Y, Bahl N, Manouvrier S, Catherine Fallet-Bianco, et al. Large spectrum Lissencephaly and pachygyria phenotypes resulting from denovo missense mutation in Tubulin 1 A (TUBA1A). Hum Mutat. 2007; 28: 1055-1064.
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JE, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELIN mutations. Nat Genetics. 2000; 26: 93-96.
- Gastout H, Pinsard N, Rayboud CH, Aicardi J, Zinkin B. Lisencephaly (Agyria-Pachygyria) clinical findings and serial EEG studies. Dev Med Child Neurol. 1987; 29: 167-180.