
Case Report
Austin J Clin Case Rep. 2021; 8(5): 1213.
Childhood Adrenocortical Carcinoma: A Case Report
Tripathi K*, Pande V and Agarkhedkar S
Department of Paediatrics, Dr DY Patil Medical College, India
*Corresponding author: Kriti Tripathi, Department of Paediatrics in Dr DY Patil Medical College, Pune, Maharashtra, India
Received: May 04, 2021; Accepted: May 26, 2021; Published: June 02, 2021
Abstract
Adrenal tumors can arise either from cortex or from medulla; both the regions being structurally and functionally different. They are categorized as functional (hormone-secreting) or silent and as either benign or malignant. Adrenocortical Carcinoma (ACC) are rare tumors that have a bimodal distribution, although most adult ACC are non-functional, in pediatric age group, nearly 95% are functional Our case report is presented because of its rarity consisting of our patient with virilising signs at the age of one and a half years.
Keywords: Childhood adrenocortical carcinoma; Pediatric cancers
Case Presentation
A one and a half year old male child presented to us with progressive virilising signs of excessive increase in the weight and the height since 10 months, deepening of voice with increase in the growth of pubic hair since 7months, genital organs and skin changes since 1 month.
On examination the child was conscious and irritable .The vitals were stable, but the blood pressure was above the 99th centile for his age (126/94mmhg).
His height was 82.5cm (+1SD), weight was 13.5kgs (SDS + 6.03). He had signs of virilization in the form of deepening of the voice, hypertrophy of the penis, increased pubic hair and skin changes with acne. There was no palpable mass in the abdomen. There was no family history of malignancy. Laboratory investigations revealed normal hematological and biochemical parameters. Thyroid profile was within normal limits .Morning (0800 hours) serum cortisol was 12μg /dL (3.7-19.40 μg/dL) night serum cortisol was 13.60 μg/dL, both within normal limits . Other hormonal levels were as follows: dihyrdro-epiandrosterone sulfate 1401.49 μg/dL (32.7-276), 17-OHprogesterone 9.41ng/mL (1.0), and testosterone 2169.62ng/mL (7- 20).
Ultrasound of abdomen revealed a solid hypoechoic lesion of 49mm in size in the right suprarenal region without calcification or cystic component with the lesion being hypovascular on doppler mode. Computed Tomography (CT) of the abdomen showed a mildly heterogenous enhancing lesion measuring 5.3 cm *3.7 cm in the right suprarenal fossa, in close relation to the liver and the superior pole of the right kidney. The right adrenal was not seen separately from the lesion. Mild mass effect on the superior pole of the right kidney was present. No obvious calcification or necrosis noted in the lesion (Figure 1 and 2).
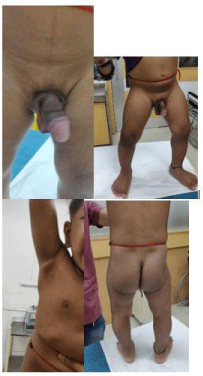
Figure 1:
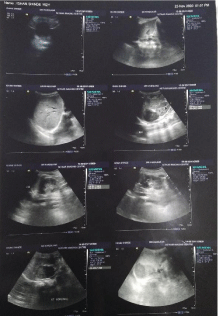
Figure 2: Computed Tomography of abdomen and pelvis without and with
contrast.
A provisional diagnosis of an adrenocortical tumor was made. Correction of general condition was done. Details about the disease, plan of management, and risks were explained to the parents. After written consent and preanesthetic checkup, surgery was undertaken under general anesthesia. Right adrenalectomy was performed with a 5.7x3.7 cm mass arising from right adrenal was removed and the mass was sent for histopathological study.
On histopathological examination, multiple regressed sections studied show tumor cells. Arranged in sheets and trabeculae individual tumor cells are round to oval to polygonal cells .Cells were pleomorphic with hyperchromatic nuclei. Cytoplasm - eosinophilic and abundant with some cells showing bubbly/clear cytoplasm. Section also show thick broad fibrous band between parenchyma. No significant mitotic activity seen. Multiple foci of necrosis seen throughout tumor. No lymphovascular or capsular invasion is seen. According to Modified Weiss criteria, a total score of 3 concluded to adrenocortical carcinoma.
However, in view of low mitotic activity and absence of atypical mitotic figures, ki67/MIB 1 was index indicated for proper evaluation. After confirming the diagnosis the patient was advised and referred to get admission in Pediatric Hematology-Oncology department for chemotherapy and further management.
Discussion
Tumors arising from the adrenal cortex are usually adrenal adenomas or Adrenocortical Carcinomas (ACCs); whereas those arising from the adrenal medulla are neuroblastomas and Pheochromocytomas (PCCs). Conventionally, they are classified into functional and nonfunctional tumors. Adrenal adenomas, ACCs, and PCCs are functional tumors, whereas neuroblastomas are nonfunctional. Adrenocortical tumors (ACTs) are rare in children, comprising <0.2% of pediatric neoplasms [1]. The ACTs have varied presentation either virilizing forms or presentation with Cushing’s syndrome, or both. In children, due to the rapid development of symptoms they come to attention early, however, if not diagnosed and treated early, It can have a downhill course. The last decade has seen the emergence of new diagnostic imaging modalities. There is also intense ongoing research in newer treatment modalities as these tumors can be unresectable or have a high recurrence rate.
ACTs contribute 6% of pediatric adrenal tumors [2]. Although the first case of ACT was reported in Lancet in 1865. Most of the children with functioning ACT, who had undergone adrenalectomy till 1937, did not survive postoperatively due to adrenocortical insufficiency [3]. Vast majority (91.8%) of pediatric ACC occurs among Caucasians and Asians are affected only in 5.9% of cases [4].
ACTs have a bimodal age distribution, with first and fifth decades of life being peaks [5]. Two genetic syndromes have a clear association with ACT: (i) Li–Fraumeni syndrome, a familial cancer syndrome having mutation of P53 gene, and (ii) Beckwith– Wiedmann syndrome having mutations in the 11p15 region, [3] Genetic alterations or the above congenital syndromes are associated with about half of the cases of ACC [6]. About 90% of the cases of ACT are functional in children in contrast to <50% cases in adults [7]. Virilization is the most common presentation of ACT seen in 80- 84 % of cases [7]. in the form of acne, pubic hair, and hypertrophy of clitoris. Cushing syndrome is the second most common presentation in childhood ACT, seen in one-third cases with moon facies, centripetal distribution of fat, and striae over the skin and plethora [3].
Surgery is the cornerstone of the treatment in ACTs. Functional ACTs are presumed to cause suppression of the contralateral adrenal gland [3]. Modified Weiss criteria is an established method for the diagnosis of ACC [8,9].
Out of nine histological criteria, three findings are required to make the diagnosis of ACC and our case fulfilled three Weiss criteria. The morphology was characteristic of ACC. No single immunohistochemistry marker is 100% sensitive or specific in ACC. Although combination chemotherapy with etoposide and cisplatin was described in the postoperative management. The role of chemotherapy is not yet established due to limited number of cases [13]. Adjuvant therapy with mitotane (dichlorodiphenyldichloroethane) is an established option in advanced and metastatic ACC in adults [11], an insecticide derivative causing adrenocortical necrosis and tumor regression. However, its role in children pediatric ACC is not well established [13]. Age at diagnosis is considered as an important prognostic factor, with children <2 years having good outcome. Children >2 years of age are reported to be having only 29% survival rate [10]. Microscopically, mitotic counts >20/50 HPF are described to be associated with poor outcome [11]. Recently, Ki-67 proliferation index has been identified as the most important prognostic factor for recurrence-free survival [12].
Conclusion
A multidisciplinary approach is needed in the management of childhood adrenal tumors, which is a challenging task. The rarity of ACTs may point toward the role of environmental and genetic factors. Moreover, childhood hypertension due to adrenal tumor is a potentially fatal condition. Surgery is the optimal treatment, and event-free survival is expected even if the age group falls within poor prognostic criteria. ACT in children can be diagnosed early due to rapid development of symptoms. The prognosis is worse if diagnosed late.
References
- Stewart JN, Flageole H, Kavan P. A surgical approach to adrenocortical tumors in children: The mainstay of treatment. J Pediatr Surg. 2004; 39: 759- 763.
- Chudler RM, Kay R. Adrenocortical carcinoma in children. Urol Clin North Am. 1989; 16: 469-479.
- Ribeiro RC, Michalkiewicz EL, Figueiredo BC, DeLacerda L, Sandrini F, Pianovsky MD, et al. Adrenocortical tumors in children. Braz J Med Biol Res. 2000; 33: 1225-1234.
- McAteer JP, Huaco JA, Gow KW. Predictors of survival in pediatric adrenocortical carcinoma: A Surveillance, Epidemiology, and End Results (SEER) program study. J Pediatr Surg. 2013; 48: 1025-1031.
- Liou LS, Kay R. Adrenocortical carcinoma in children. Review and recent innovations. Urol Clin North Am. 2000; 27: 403-421.
- Ribeiro RC, Fighueiredo B. Childhood adrenocortical tumors. Eur J Cancer. 2004; 40: 1117-1126.
- Gundgurthi A, Kharb S, Dutta MK, Garg MK, Khare A, Jacob MJ, et al. Childhood adrenocortical carcinoma: Case report and review. Indian J Endocrinol Metab. 2012; 16: 431-435.
- Lau SK, Weiss LM. The Weiss system for evaluating adrenocortical neoplasms: 25 years later. Hum Pathol. 2009; 40: 757-768.
- Aubert S, Wacrenier A, Leroy X, Devos P, Carnaille B, Proye C, et al. Weiss system revisited: A clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am J Surg Pathol. 2002; 26: 1612-1619.
- Ciftci AO, Senocak ME, Tanyel FC, Büyükpamukçu N. Adrenocortical tumors in children. J Pediatr Surg. 2001; 36: 549-554.
- Libé R. Adrenocortical Carcinoma (ACC): Diagnosis, prognosis, and treatment. Front Cell Dev Biol. 2015; 3: 45.
- Beuschlein F, Weigel J, Saeger W, Kroiss M, Wild V, Daffara F, et al. Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab. 2015; 100: 841-849.
- Cho MJ, Kim DY, Kim SC, Kim TH, Kim IK. Adrenocortical tumors in children 18 years old and younger. J Korean Surg Soc. 2012; 82: 246-250.