
Research Article
Austin J Clin Case Rep. 2021; 8(7): 1221.
Screening for HNF1B Gene Mutations, in Four People with Typical Diabetes MODY5 Clinical Characteristics
Alejandro de Dios1, Sofia Irene Trobo2, Anette Marianne Prior Gjesing3, Torben Hansen3, Gustavo Daniel Frechtel1 and Ariel Pablo López1,2*
¹University of Buenos Aires, Hospital de Clínicas “José de San Martín”, Genetics Division, Clinical Hospital, School of Medicine, Argentina
²University of Buenos Aires, Molecular Genetics, School of Pharmacy and Biochemistry, Argentina
³University of Copenhagen. Novo Nordisk Foundation Center for Basic Metabolic Research, Section of Metabolic Genetics, Faculty of Health and Medical Sciences, Denmark
*Corresponding author: Ariel Pablo Lopez, University of Buenos Aires, School of Pharmacy and Biochemistry, Novo Nordisk Foundation Center for Basic Metabolic Research, Section of Metabolic Genetics, Faculty of Health and Medical Sciences, Av Cordoba 2351, 4to piso sala 5; Hospital de Clinicas “José de San Martín”, CABA, Argentina
Received: May 18, 2021; Accepted: June 21, 2021; Published: June 28, 2021
Abstract
Introduction: people with clinical characteristics of MODY benefit with a correct genetic diagnosis and are often only studied if having family history. In this work there were studied four people selected per their clinical characteristics of genitourinary abnormalities and MODY Diabetes, using the worldwide inclusion criteria for MODY5 except for a family history of diabetes or kidney disease. Methods: gene mutation screening in four people with clinical characteristics of MODY5 in search for alterations in the HNF1B gene with Sanger or NGS sequencing, and bioinformatic tools to analyze the results of the sequences.
Results: from four people studied we found three mutations in the HNF1B gene, including a missense mutation previously described and two de novo whole gene deletions. The other person did not present any alteration in that gene even having clinical characteristics.
Conclusions: people with clinical characteristics of MODY and having pancreatic, renal, kidney or genital located abnormalities are candidates for genetic screening of HNF1B. Yet, genetic screening of HNF1B should not only be restricted to such people but should also be considered in people without diabetes but having those other characteristics. We suggest also, the study of people even in the absence of family history, given that the possibility of occurrence of de novo mutations is underestimated.
Keywords: Nephropathy; Monogenic diabetes; MODY 5; HNF1B gene; De novo mutations
Introduction
Known monogenic forms of diabetes are caused by genetic alterations leading to specific clinical characteristics including diabetes. Among monogenic forms of diabetes is Maturity-Onset Diabetes of the Young (MODY) where 14 different causal genes have been described (OMIM #606391) [1,2]. The genetic alterations involved in MODY are located in genes affecting pancreatic development and function, and depending on the location of the mutation, synthesis and/or secretion of insulin are altered [3,4].
MODY people are usually characterized by a positive family history of diabetes with an autosomal dominant inheritance, an early age at diagnosis typically before 25 years, a non-insulin-dependent clinical presentation and absence of anti-ß-cell antibodies.
Mutations in six genes are considered to be the most frequent cause of MODY. MODY1, MODY2 and MODY3 are caused by heterozygous mutations in the hepatocyte nuclear factor 4 alpha gene, the glucokinase gene and the hepatocyte nuclear factor 1 alpha gene respectively, whereas MODY4, MODY5 and MODY6 are caused by mutations in the IPF1 gene, in the hepatocyte nuclear factor 1 beta gene (HNF1B) and in the NEUROD1 gene respectively [1].
Hepatocyte nuclear factor 1B (HNF1B) gene is located on chromosome 17q21.3, has 9 exons and encodes a 557-amino-acid protein. Its structure is characterized by a highly-conserved DNA binding domain. It encodes a transcription factor that forms a twomolecule homodimer or a heterodimer with HNF1A protein with which it is structurally related [5]. In addition, it is known for its role in tissue-specific gene expression in several organs, including the liver, kidney, and pancreatic islets and genital tract, and it is involved in the ß-cell transcription factor network [5,6]. HNF1B is widely distributed and essential for embryonic survival. Early expression of HNF1B has been observed in the kidney, liver, bile ducts, thymus, genital tract, pancreas, lung, and gut embryonic tissues [7,8].
The first HNF1B mutation was described by Horikawa et al. in 1997, but despite its initial identification as a diabetes disease gene, mutations in HNF1B/MODY5 are an infrequent cause of MODY, accounting for <2% of cases, compared with approximately 40% attributed to GCK/MODY2 [9,10].
A wide spectrum of HNF1B mutation phenotypes has been reported, as well as an enormous variability of the disease severity even within families. Clinical features observed in people with HNF1B mutations, are closely related to the expression profile of HNF1B, and HNF1B (MODY5) people encompasses a clinical spectrum comprising diabetes, pancreas atrophy with subclinical exocrine deficiency, progressive nondiabetic nephropathy, kidney and genital malformations, and liver abnormalities [9-11]. Consistent with the important role of HNF1B in pancreatic development, pancreatic malformations have been described in mutation carriers [12,13].
The renal disease described is very heterogeneous yet always due to aberrant renal development and includes: renal cysts, familial hypoplastic glomerulocystic kidney disease (GCKD), renal malformations (for example, single and horseshoe kidney), and atypical familial hyperuricaemic nephropathy [14-17].
The spectrum of severity can vary, ranging from solely MODY or kidney involvement to multiorgan disease, it is often of early-onset, and insulin treatment is usual in MODY5 people [10,17]. People with mutations in HNF1B have impaired insulin secretory responses to glucose and insulin secretagogues and show progressive loss in basal insulin secretion [18,19].
Although HNF1B mutations are not usually associated with diabetes in childhood, single cases with onset as early as the neonatal age have been described [16,20]. However, MODY5 typically manifests in the third or fourth decade of life and is seen in approximately half of adults with HNF1B mutations [18].
Mutations in HNF1B are inherited in an autosomal dominant pattern, although up to 50% of mutations occur de novo. The most predominant alterations include missense/nonsense, small deletions and gross deletions with the latter accounting for approximately a quarter of the mutations [21-23]. Functional studies have shown mutations with either loss of function, dominant negative actions, or gain of function [16].
In the present work, we describe the screening of mutations in four people with clinical characteristics of MODY5 in search for alterations in the HNF1B gene.
Materials and Methods
Four people were selected according to their clinical characteristics to undergo genetic screening. The inclusion criteria were: 1) Diabetes or dysglycemia diagnosed before 40 years of age, 2) presence of genitourinary malformations, 3) detectable levels of C-peptide, 4) absence of beta cell autoimmunity and 5) family history of diabetes or kidney disease. Nevertheless, the absence of family history was not an exclusion criteria because a high percentage of mutations arise de novo as described in the introduction.
Genomic DNA of the selected people was extracted from peripheral lymphocytes using the MagNA Pure system (Roche, Basilea, Switzerland), followed by quantification using a DeNovix DS-11 FX+ Spectrophotometer (DeNovix Inc., Wilmington, USA).
DNA in the targeted region which included the coding regions and exon/intron boundaries of HNF1B gene was captured and sequenced using the Illumina HiSeq2000 Analyzers (Illumina, California, U.S.A.) as described in Gao R, Liu Y, Gjesing AP, et al. [24]. All coding regions were covered with a minimum mean depth of 20X. Qualified reads were aligned to the reference human genome (UCSC hg19) using Burrows-Wheeler Alignment Tool (http://biobwa. sourceforge.net) and SNPs and indels were identified using GATK (https://www.broadinstitute.org/gatk/).
Variants selected were located in coding regions and were considered putative MODY-variants if they were non-synonymous, if MAF in public databases (dbSNP: http://www.ncbi.nlm.nih.gov/ SNP/; 1000G: http://browser.1000genomes.org/index .html and EXAC: http://exac.broadinstitute.org/) were below 1% and were predicted functional using the Condel prediction program (www.bg.upf.edu).
Person 1
A 39-year-old man under study for chronic liver and kidney disease with no etiological diagnosis at inclusion into the study. He presents a family history with his mother having solitary kidney, a history of diabetes and hepatic impairment function. She died at the age of 50 years being treated with hemodialysis.
The person has a single kidney through congenital absence of the right kidney. He has multiple cysts on the left kidney and recurrent kidney stones with normal urinary metabolic parameters. He was diagnosed with chronic kidney disease (stage III b) at the age of 20 years and with hypothyroidism and diabetes at the age of 19 years. He presents a glucose level of 900 mg/dl without ketosis. He was diagnosed as type 1 diabetes with AGAD and ICA negative auto antibodies. He is currently receiving insulin treatment without having retinopathy or neuropathy but with chronic high levels of transaminases (normal liver biopsy).
Complementary studies: Abdominal computed tomography: lithiasis 10 x 10 mm in diameter in the left kidney calyx. Multiple calcifications related to the head of the pancreas, hypotrophic appearance. Liver shape, size and structure preserved (Figure 1).
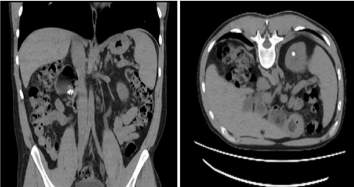
Figure 1: Person 1 CT scan showing the absence of both the right kidney and
urinary tract and urolithiasis in the left kidney calyx.
Current laboratory results: Creatinine: 3.47 mg/dl, total bilirubin: 1 mg/dl, GOT: 115 IU/L, ALT: 239 IU/L, C peptide: 0.2 ng/ml, glucose: 115 mg/dl and HbA1c: 36 mmol/mol (5,4%). (Normal values: creatinine: 0.7 to 1.3 mg/dl for men; total bilirubin: 0.3 to 1.9 mg/dl; GOT: 5 a 40 UI/L; ALT: 7 a 56 IU/L; C peptide: 0.5 to 2.0 ng/ mL; glucose: 70 to 110 mg/dl; HbA1c: less than 39 mmol/mol (5,7%)).
Current medication: Levothyroxine 100ug, intensified insulin therapy in basal-bolus regimen (NPH insulin plus aspart: 0,5UI/kg/ day), Candesartan 8mg.
We suspected MODY5 due to the presence of genitourinary abnormalities, pancreatic atrophy, and diabetes onset at early age without evidence of autoimmunity or increased liver enzymes.
Person 2
A 41-years-old man, with diabetes diagnosed at the age of 40 years. He presented polycystic kidney disease with normal renal function. He has no familiar history of diabetes or kidney disease.
Complementary studies: Renal ultrasonography: bilateral renal cysts between 5 and 45 mm, with no other pathological data.
Laboratory results at DM diagnosis: He had a glycaemia of 300 mg/dl and was taking steroids for back pain. Regarding his body mass index (BMI), at the time of the query to our Hospital, he was overweighed (BMI: 28.08).
Current Laboratory results: Creatinine: 1,32 mg/dl (MDRD >60ml/min), total bilirubin: 1,1 mg/dl, GOT: 45IU/L, ALT: 67IU/L, C peptide: 1.2ng/ml, glucose: 123mg/dl and HbA1c: 39 mmol/mol (5,7%). No evidence of beta cell autoimmunity: aGAD, ICA and IAA negative. (Normal values: creatinine: 0.7 to 1.3 mg/dl for men; total bilirubin: 0.3 to 1.9 mg/dl; GOT: 5 a 40 UI/L; ALT: 7 a 56 IU/L; C peptide: 0.5 to 2.0 ng/mL; glucose: 70 to 110 mg/dl; HbA1c: less than 39 mmol/mol (5,7%)).
Current medication: He has begun a treatment with metformin 1700mg/day and linagliptin 5mg/day and due to that, he is improving glycemic control and weight (last measure of BMI: 25).
We suspected MODY5 due to the presence of genitourinary abnormalities, diabetes onset at early age without evidence of autoimmunity or increased liver enzymes.
Person 3
A 14-year-old man with chronic renal insufficiency and bladder malformation. He was evaluated for short stature. He has an obese mother diagnosed with diabetes at 48 years of age, to date treated with insulin. His sister is 16 years old having obesity and being under treatment with metformin.
Complementary studies: Renal ultrasound: hypotrophic bladder with bilateral pyelocaliceal dilatation.
Current laboratory results: In biochemical studies, dysglycemia was found in this person. The oral glucose tolerance test presented: fasting glucose 88mg/dl, glucose at 120 minutes: 161mg/dl with insulinemia of 2.6µUI/ml (fasting state) and 47.7µUI/ml (al 120 minutes). Other data: Hematocrit: 33%, hemoglobin: 11,8gr/dl, creatinine: 2,3mg/dl, normal liver function tests, HbA1c: 37 mmol/ mol (5,5%) and fructosamine: 295µmol/L. (Normal values: OGTT, fasting: less than or equal to 100 mg/dl, 120 min: less than 140 mg/ dl; hematocrit: 40.7% to 50.3% for men; hemoglobin: 13.8 to 17.2 g/dl for men; creatinine: 0.7 to 1.3 mg/dl for men; total bilirubin: 0.3 to 1.9 mg/dl; GOT: 5 a 40 UI/L; ALT: 7 a 56 IU/L; C peptide: 0.5 to 2.0 ng/ mL; glucose: 70 to 110 mg/dl; HbA1c: less than 39 mmol/mol (5,7%); fructosamine: 200-285 µmol/L).
Current medication: Enalapril 2,5mg/day, sodium bicarbonate, Vitamin D, folic acid and ferrous sulfate.
MODY5 was suspected due to the presence of genitourinary abnormalities and dysglycemia onset at early age without evidence of autoimmunity or family history of DM.
Person 4
A 28-year-old woman with a diagnosis of Diabetes during Ovarian Cyst Surgery, with a personal history including: renal cysts, right renal agenesis, pancreatic hypoplasia, bicorne uterus. Without familiar history of Diabetes or Kidney disease.
Onset clinical characteristics: BMI 22.06 (weight 53Kg; high: 155cm) A1c at diagnosis: 7.20%; C peptide: 2.7 ng/ml.
Treatment during hospitalization: Insulin therapy, current treatment: Vildagliptin 50 mg / day; current metabolic control: A1c 7.1; BMI 22.55.
We suspected MODY5 due to the presence of genitourinary abnormalities, diabetes onset at early age without evidence of autoimmunity or increased liver enzymes.
Results
We found a probable MODY5 causing mutation in Person 1. The mutation has previously been described [16]. A heterozygous whole gene deletion was identified in Person 2 and person 4 which are causal of MODY5. In their families, there are no antecedents of diabetes or renal pathology, which strongly indicates that those are de novo mutations. In Person 3, there were no mutations suspicious of being causal of MODY5.
Person 1: gene HNF1B, c.1021G>A
In this person, a missense mutation was found in exon 4 of HNF1B, resulting in a substitution of Arg to Gln at position 276 (NM_000458.2, c.1021G>A). This variant has not previously been found in public databases and was predicted deleterious. In addition, it has previously been found in a family having diabetes as well as renal cysts [16]. Thus, this variant is very likely a causal MODY5 mutation. In this person, we found no indication of any insertion or deletion.
Person 2: gene HNF1B, heterozygous whole gene deletion
In this person, there were no MODY-like mutations in the coding region +/- 3 nucleotides from exon/intron boundaries. However, we found that the depth of all exons in HNF1B is half the depth of the remaining samples in the same sequencing lane. This is a strong indication that there is a heterozygous whole gene deletion of the HNF1B in this person.
Person 3: gene HNF1B, no mutation found
In this person, no MODY-like mutations in the coding region +/- 3 nucleotides from exon/intron boundaries were found in HNF1B gene. Nor was there any indication of insertions or deletions based on the depth.
Person 4: gene HNF1B, heterozygous whole gene deletion
In this person, there were no MODY-like mutations in the coding region +/- 3 nucleotides from exon/intron boundaries. However, we found that the depth of all exons in HNF1B is half the depth of the remaining samples in the same sequencing lane. This is a strong indication that there is a heterozygous whole gene deletion of the HNF1B in this person.
Discussion
Since the identification that mutations in HNF1B can cause MODY, over 170 mutations have been described. People with HNF1B-related disorders have been reported with wide spectrum phenotypic characteristics. Nevertheless, renal manifestations clearly appear to be the most common, followed by pancreatic and genital abnormalities. Neurological complications appear to be restricted to people with deletions in the 17q12 region [22].
In this work, we report the results of the study of four people with different personal and familial characteristics including two without a familial history of diabetes, renal or kidney disease. In the present study, Person 1 had familial history of diabetes, with liver and kidney disease and presented a mutation very likely to be the cause of his alterations. This variant is located in the DNA binding domain of the protein and has previously been reported to co-segregate with the symptomatology within a family. Thus, we assume it is the causal mutation. The symptoms reported previously are similar but not equal to the symptoms presented in this study. Our person presented a more severe clinical profile, and his mother had different symptoms and died of complications related to her disease. Therefore, our findings indicate that the same mutation can cause diverse abnormalities in different individuals [16].
Persons 2 and 4 had an entire deletion of one allele of the HNF1B gene. Thus, as previously published, those deletions can be asserted as causal of the diabetes and other symptoms in both Persons. In addition, they have no family history of diabetes or kidney disease so we speculate that the alterations found represents de novo mutations [23].
And finally, Person 3 has a personal history of diabetes and renal abnormalities and a familial history of diabetes only, but surprisingly no MODY5 related mutation was found. This finding suggests that there may be coexisting alterations within this family that can cause the renal insufficiency in the proband while another alteration is causal of the extra diabetic characteristics in other family-members.
Each person shows a particular phenotype making ascertainment of association or suspicion of MODY5 very difficult. Moreover, there is little data regarding complication, prevention or treatment [22].
The abnormal morphogenesis of the pancreas is likely one of the causes of diabetes in MODY5 people, but another suggested molecular mechanism is related to altered GLUT2 expression that occurs mainly in hepatocytes, renal proximal tubule cells, and pancreatic β-cells [18]. Mutant HNF1B proteins have a defect in DNA binding and limited ability to increase the transcription of GLUT2, which plays a primary role in β-cell insulin secretion, as it senses external glucose and transports it into pancreatic β-cells. In vivo studies showed that GLUT2 deficiency results in diabetes with impaired glucose sensing by pancreatic β-cells [25-27]. So, decreased expression of GLUT2 in β-cells, combined with a gradual reduction in insulin secretion due to pancreas malformation, are likely involved in the pathogenesis of diabetes mellitus in MODY5 people. In addition, GLUT2 may also affect β-cell differentiation in the development of the pancreas [28].
MODY5 has been associated with either mutations within HNF1B gene or allelic deletion of chromosome 17q12 including the HNF1B locus. HNF1B locus deletion was detected in approximately 30% of adult HNF1B/MODY5 cases, and it appears to be much more frequent in cases that are diagnosed during childhood [26]. In addition, according to Chen et al., renal structural anomalies are relatively less frequent when missense mutations occur compared to other types of mutations whereas the percentage of diabetes mellitus and the frequency of insulin treatment tended to be higher [29]. It has been established in previous studies that 70% of people presenting a clinical phenotype consistent with MODY5, associated with either abnormalities of kidney morphology or impaired renal function, are carriers of HNF1B molecular alterations [23].
In conclusion, people with clinical characteristics of MODY and having pancreatic, renal, kidney or genital located abnormalities are candidates for genetic screening of HNF1B. Yet, genetic screening of HNF1B should not only be restricted to such people but should also be considered in people without diabetes having other of the above characteristics described.
Ethical Declaration
This work has been approved by the Institutional Review Committee and written informed consent was obtained from all individuals involved or from responsible family members after full explanation of the purpose and nature of all procedures used.. All human investigations were conducted according to the principles expressed in the Declaration of Helsinki as revised in 1983.
Conflicts Interest
We wish to confirm that there are no known conflicts of interest associated with this publication and there has been no financial support for this work that could have influenced its outcome.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Acknowledgement
We wish to thank all the participants and families who collaborated in any form to the development of these studies and also Rocio Sidera for her support in correcting this article.
Funding
This work was supported by grants belonging to University of Buenos Aires, Buenos Aires, Argentina and from the Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark. Both institutions had no participation in study design; in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the article for publication.
References
- American Diabetes Association (ADA). Diagnosis and classification of diabetes mellitus. Diabetes Care. 2008; 31: S55-S60.
- MIM #606391; Maturity Onset Diabetes of the Young; MODY;
- Velho G, Froguel P. Genetic, metabolic and clinical characteristics of maturity onset diabetes of the young. Eur J Endocrinol. 1998; 138: 233-239.
- Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001; 345: 971-980.
- Mendel DB, Hansen LP, Graves MK, Conley PB, Crabtree GR. HNF-1 alpha and HNF-1 beta (vHNF-1) share dimerization and homeo domains, but not activation domains, and form heterodimers in vitro. Genes Dev. 1991; 5: 1042-1056.
- Servitja JM, Ferrer J. Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia. 2004; 47: 597-613.
- Coffinier C, Barra J, Babinet C, Yaniv M. Expression of the vHNF1/HNF1beta homeoprotein gene during mouse organogenesis. Mech Dev 1999; 89: 211- 213.
- Reber M, Cereghini S. Variant hepatocyte nuclear factor 1 expression in the mouse genital tract. Mech Dev 2001; 100: 75-78.
- Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. 1997; 17: 384-385.
- Ryffel GU. Mutations in the human genes encoding the transcription factors of the Hepatocyte Nuclear Factor (HNF)1 and HNF4 families: functional and pathological consequences. J Mol Endocrinol. 2001; 27: 11-29.
- Bellanne-Chantelot C, Chauveau D, Gautier JF, Dubois-Laforgue D, Clauin S, Beaufils S, Wilhelm JM, Boitard C, Noel LH, Velho G, Timsit J: Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med. 2004; 140: 510-517.
- Haldorsen IS, Vesterhus M, Raeder H, Jensen DK, Sovik O, Molven A, et al. Lack of pancreatic body and tail in HNF1B mutation carriers. Diabet Med. 2008; 25: 782-787.
- Body-BechouD, LogetP, D’HerveD, LeFiblecB, GrebilleAG, Le Guern H, et al. TCF2/HNF-1beta mutations: 3 cases of fetal severe pancreatic agenesis or hypoplasia and multicystic renal dysplasia. Prenat Diagn. 2014; 34: 90-93.
- Carbone I, Cotellessa M, Barella C, Minetti C, Ghiggeri GM, Caridi G, Perfumo F, Lorini R. A novel hepatocyte nuclear factor-1beta (MODY-5) gene mutation in an Italian family with renal dysfunctions and early-onset diabetes. Diabetologia. 2002; 45: 153-154.
- Bingham C, Ellard S, van’t Hoff WG, Simmonds HA, Marinaki AM, Badman MK, et al. Atypical familial juvenile hyperuricemic nephropathy associated with a hepatocyte nuclear factor-1beta gene mutation. Kidney Int. 2003; 63: 1645-16451.
- Edghill EL, Bingham C, Ellard S, Hattersley AT. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet. 2006; 43: 84-90.
- Adalat S, Bockenhauer D, Ledermann SE, Hennekam RC, Woolf AS. Renal malformations associated with mutations of developmental genes: messages from the clinic. Pediatr Nephrol. 2010; 25: 2247-2255.
- Pearson ER, Badman MK, Lockwood CR, Clark PM, Ellard S, Bingham C, Hattersley AT. Contrasting diabetes phenotypes associated with hepatocyte nuclear factor-1alpha and -1beta mutations. Diabetes Care. 2004; 27: 1102- 1107.
- Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1Bassociated renal and extra-renal disease-an expanding clinical spectrum. Nat Rev Nephrol. 2015; 11: 102-112.
- Yorifuji T, Kurokawa K, Mamada M, Imai T, Kawai M, Nishi Y, et al. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: Phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1beta gene due to germline mosaicism. J Clin Endocrinol Metab. 2004; 89: 2905- 2908.
- Eun Ky Kim, Ji Seon Lee, Hae Il Cheong, Sung Soo Chung, Soo Heon Kwak, Kyong Soo Park. Identification and Functional Characterization of P159L Mutation in HNF1B in a Family with Maturity-Onset Diabetes of the Young 5 (MODY5) Genomics Inform. 2014; 12: 240-246.
- Detlef Bockenhauer & Graciana Jaureguiberry. HNF1B-associated clinical phenotypes: the kidney and beyond. Pediatr Nephrol.
- Christine Bellanne´-Chantelot, Se´verine Clauin, Dominique Chauveau, Philippe Collin, Miche` le Daumont, Claire Douillard, et al. Large Genomic Rearrangements in the Hepatocyte Nuclear Factor-1 (TCF2) Gene Are the Most Frequent Cause of Maturity-Onset Diabetes of the Young Type 5. 2005; 54: 3126-3132.
- Gao R, Liu Y, Gjesing AP, Hollensted M, Wan X, He S, et al. Evaluation of a target region capture sequencing platform using monogenic diabetes as a study-model. BMC Genet. 2014; 15: 13.
- Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE, Ellard S. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat. 2013; 34: 669-685.
- Raile K, Klopocki E, Holder M, Wessel T, Galler A, Deiss D, et al. Expanded clinical spectrum in hepatocyte nuclear factor 1b-maturity-onset diabetes of the young. J Clin Endocrinol Metab. 2009; 94: 2658-2664.
- Guillam MT, Hümmler E, Schaerer E, Yeh JI, Birnbaum MJ, Beermann F, et al. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet. 1997; 17: 327-330.
- Haumaitre C, Barbacci E, Jenny M, Ott MO, Gradwohl G, Cereghini S. Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc Natl Acad Sci USA. 2005; 102: 1490-1495.
- Chen YZ, Gao Q, Zhao XZ, Bennett CL, Xiong XS, Mei CL, et al. Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chin Med J (Engl). 2010; 123: 3326-3333.