
Research Article
Austin J Clin Immunol. 2014;1(1): 1004.
Lipopolysaccharide-responsive beige-like anchor (LRBA), a novel regulator of human immune disorders
Jia-Wang Wang1* and Richard F Lockey1,2
1Department of Internal Medicine, University of South Florida Morsani College of Medicine, USA
2Department of Internal Medicine, University of South Florida College of Medicine, James A. Haley Veterans’ Hospital, Tampa, USA
*Corresponding author: Jia-Wang Wang, Department of Internal Medicine, University of South Florida Morsani College of Medicine, USA.
Received: January 04, 2014; Accepted: January 29 , 2014; Published: February 03, 2014
Abstract
Lipopolysaccharide (LPS)-responsive beige-like anchor (LRBA) is a novel gene essential for the normal function of the immune system. It is the eighth common variable immunodeficiency (CVID) gene, mutation of which causes CVID and autoimmunity, and is associated with inflammation. LRBA is a unique CVID gene when compared to other CVID genes: it is a large, PKA anchor and vesicle trafficking regulator. Other seven CVID genes that are associated with CVID are cell receptors. However, the molecular mechanism by which LRBA regulates the immune system is unknown. LRBA protein contains Concanavalin A (ConA)-like lectin binding domain, Vacuolar protein sorting-27, hepatocyte growth factor-regulated tyrosine kinase substrate domain and signal transducing adaptor molecule (VHS) domain, two RII binding motifs, WD-like (WDL), and Beige and Chediak-Higashi (BEACH) domains and five WD40 repeats (WBW super domain). An LC3 interaction region (LIR), which is involved in autophagy, is also predicted in LRBA. The WBW super domain defined the WBW gene family, members of which appear to function as scaffolding proteins in vesicle trafficking and are important in human diseases. The cargo proteins regulated by LRBA through vesicle trafficking may include cytokines and antibodies for secretion, plasma membrane proteins for disposition on the membrane, and proteins trafficking between different membrane compartments, or proteins for degradation through lysosome/autophagy or proteasome degradation. Specifically, LRBA interacts with multiple important signal transduction pathways, including epidermal growth factor receptor (EGFR), Notch, PKA, Ras, E2F1, p53, and mitogen-activated protein kinases (MAPKs). These molecular interactions may help to understand why and how LRBA is involved in critical cellular processes such as cell proliferation, apoptosis and autophagy, and plays a fundamental role in the normal immune system.
The discovery of the LRBA gene
To discover genes responsible for B cell development, a short DNA sequence (143bp), 7a65, was obtained through a gene trapping method that requires the fusion of Escherichia coli lactose operon lacZgene and a cellular gene at both the transcriptional and translational levels, in order for the cell to express ß-galactosidase [1]. To obtain the full length sequence of the transcript, primers were designed from this sequence and PCR fragments were obtained from a mouse B lymphocyte cDNA library. The 5’ and 3’ rapid amplification of cDNA ends (RACE) techniques were further used to obtain the full length murine Lrba gene sequence from murine cell lines as well as from the liver and thymus of C57BL6/J mice [2]. A search of the GenBank found that the murine Lrba gene has a high degree of homology to a 7.3-kb human partial cDNA sequence called Beige- Like Protein (BGL) [3], which belongs to the human LRBA gene. The rest of the cDNA sequences of human LRBA were obtained from human lung, brain, and kidney cDNA libraries to complete the human LRBA sequence [2]. Human LRBA and murine Lrba proteins are 90% identical (2587/2859)with 94% positive(2690/2859) amino acid homology. Three murine Lrba isoforms with differences at the C-terminal were identified [2], while the human LRBA has two major isoforms [2,4]. LRBA has structural similarity to the lysosomal trafficking regulator (LYST) and is potentially an A-kinase anchoring protein (AKAP) for it has two regulatory subunit (RII) binding motifs to bind the RII subunit of cAMP-dependent protein kinase [2]. It belongs to the WDL-BEACH-WD40 (WBW) gene family [2] and is colocalized with the Golgi complex (GC), lysosomes, endoplasmic reticulum (ER), plasma membrane, and perinuclear ER as demonstrated by GFP fluorescence confocal and immunoelectronic microscopy. This is the first direct evidence that a WBW family protein can physically associate with various vesicular compartments in cells [2]. LRBA is also associated with motor proteins involved in vesicle trafficking [5,6]. LRBA-deficient B cells show abnormally high numbers of GCs [4]. It too is over expressed in several different cancers and its promoter activity is inhibited by p53 and increased by E2F1 [7]. Repression of LRBA expression by RNA interference, or a dominant-negative mutant, down-regulates the phosphorylation of epidermal growth factor receptor (EGFR) and significantly inhibitscancer cell growth [7]. Three papers published in 2012 demonstrate that deleterious mutations of LRBA cause CVID and autoimmunity, and are associated with inflammation. LRBA deficient patients have an early onset of more severe and potentially life-threatening CVID [4,8-10], demonstrating that LRBA plays a fundamental role in the normal immune system.
Clinical features of LRBA deficiency
CVID is the most common late onset primary immunodeficiency disease (PID) and is characterized by hypogammaglobulinemia and recurrent bacterial infections [11]. It is caused by defective B cell differentiation and impaired secretion of immunoglobulins [12,13]. CVID is a diagnosis of exclusion and is highly heterogeneous, genetically, immunologically and clinically [14-16]. About twothird of CVID subjects have an autoimmune problem [17], most commonly autoimmune hemolytic anemia (AHA), autoimmune thrombocytopenia, rheumatoid arthritis, and pernicious anemia [18]. The etiology of about 80% of CVID remains unknown [18], although over the past ten years, significant progress has been made in elucidating genetic mechanisms that result in a CVID phenotype. A small group of genes are found to be associated with or cause CVID [15]. These include the members of the B cell coreceptor complex (CD19 [19] , CD21 [20] and CD81 [21]), CD20 [22], transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) [23] and B cell-activating factor receptor (BAFFR) [24], and inducible costimulator (ICOS) [25]. The discoveries of CVID-causing mutations of these genes show that a monogenic defect may produce the whole spectrum of CVID, and that it is possible to unravel the genetic causes underlying most human diseases thought to be polygenic [26].
All of the above named receptors are involved in the stimulation, survival and development of B cells. CD20 is expressed on all stages of B cell development, except for early pro-B and plasma cells. CD21 facilitates internalization of immune complexes by B cells to enhance antigen presentation [27]. It forms a B cell membrane complex with CD19 and CD81 to augment the B-cell receptor response to antigen [27]. Both BAFFR and TACI play an important role in B-cell biology and development [28]. BAFFR is a critical B cell survival gene, disruption of which causes a dramatic drop in B cell numbers [29,30]. BAFF is a ligand for both BAFFR and TACI. Abnormally active BAFF signaling may play a role in autoimmunity. Likewise, excessive BAFF plays a role in promoting an autoimmune condition in mice which closely resembles systemic lupus erythematosus (SLE) in humans. TACI is a negative regulator of BAFF signaling in B-cell survival and responses [31], is important for switched memory B cells [32] and is associated with autoimmunity [23,33]. TACI deficient mice have increased immune globulin production and autoimmunity [34]. However, some CVID receptor mutations identified in CVIDs are present in healthy individuals; therefore, the same mutations may have a great variability in disease presentation, indicating that other underlying factors are also involved in this disease. The importance of these receptors is illustrated by the fact that they are therapeutic targets for human diseases. For example, anti-CD20 therapy is used extensively to treat B cell lymphoma and multiple sclerosis [35]. A therapeutic monoclonal antibody against BAFF is the first SLE medication approved by the FDA in over 40 years to treat this disease [36].
LRBA is significantly different from the other seven CVID genes (Table 1): First, it encodes a 319 kD large protein composed of multiple domains [1,2] and could serve as a scaffold to interact with multiple proteins. Other CVID proteins are relatively small, from 19 kD to 145 kD. Second, the seven CVID genes are B cell membrane receptors, except for ICOS which is on T cells, while LRBA is ubiquitously expressed as a vesicle trafficking regulator, required for homeostasis and activation of plasma membrane receptors [2,37]. Thus, LRBA may regulate other CVID genes, for example, CD19, CD20 and BAFFR, because their levels are low when LRBA is absent [4]. Third, LRBA deficiency causes both immunodeficiency and autoimmunity [4,8,38]. All 11 LRBA deficient CVID subjects identified thus far have autoimmune diseases (Table 4). TACI mutations also are associated with autoimmunity but to a lesser degree (36% vs. 23% of patients with wild-type TACI) [39]. Finally, LRBA is the only CVID protein that is a protein kinase A(PKA)anchor and thus can function as PKA to regulate protein activity by phosphorylation. In addition to these above unique features, LRBA also is unique in regulating autophagy, apoptosis, membrane dynamics and receptor signaling, all of which are important for inflammation [1,2].
![]()
CVID genes
Molecular weight (kD)
Sub cellular localization
Function
Diseases
ICOS: inducible co stimulator
27
T Cell Surface
Germinal center formation, isotype class switching, and the development of memory B cells
CVID [26]
TACI: transmembrane activator and calcium modulator and cyclophilin ligand interactor
32
B Cell Surface
Mutations impair the development of IgA- and IgG-secreting plasma cells and promote lymphoproliferation. Antibody class switching autoimmunity. Negative regulator of B-cell[34,73 ]
Significantly reduced IgA levels [33] mutated in 5% -10% of CVID patients [23] Common variable immunodeficiency and IgA deficiency[23,33 ]
CD19
61
B Cell Surface
B cell co-receptor in conjunction with CD21 and CD81
CVID [19]
BAFFR: B cell activating factor belonging to the TNF family receptor
19
B Cell Surface
B-lymphocyte survival depends on BAFF-R signaling because BAFF-R deficiency blocks B-cell development at the stage of transitional B cells. The most important B cell survival signals [74]
An adult-onset antibody deficiency syndrome [24] High levels of BAFF in mice, lead to an autoimmune disease similar to SLE [75]
CD81
26
B Cell Surface
Regulation of cell development, activation, growth and motility
CVID [21]
CD21
145
B Cell Surface
Receptor for Epstein-Barr virus (EBV) binding on B and T lymphocytes Expansion of CD21lo in CVID patients has been clearly associated with a higher incidence of splenomegaly [39] and more recentlywith autoimmune cytopenia [7]
CVID [20]
CD20
35
B Cell Surface
Impaired T cell-independent antibody responses22markedly augment antigen presentation and the B-cell receptor response to antigen [27] development and differentiation of B-cells into plasma cells
Impaired T cell-independent antibody responses [22] Expansion of CD21lo has been found in patients with SLE [76] and CVID [77]
LRBA
320
Golgi apparatus, nucleus, plasma membrane, and cytoplasm
Vesicle trafficking regulator required for other CVID genes
Switched memory B cells are low[4,8,38 ] Disrupting BAFFR signaling causes a dramatic drop in B cell numbers [30] More CD20 positive B cells and respond to anti-CD20 therapy[4,8,9 ]
Table 1: Comparison of eight CVID genes.
![]()
Gene
LIR sequence
Length
LIR-TP53INP1
EKEDDEWILVDFI
13
LIR-LRBA
EEEDDDYVELKVE
13
LIR-TP53INP2
EDEVGDWLIIDLP
13
Homology
*.* .::: :..
Table 2: The potential LIR of LRBA has high homology with two known LIR motifs.
![]()
Clinical conditions
Manifestations
Patient 1-5 from the study [4]
Patient 1
Autoimmune
Idiopathic thrombocytopenic purpura (ITP)
Treatment
Intravenous immunoglobulin (IVIG) replacement
Immunologic investigations
low immunoglobulin levels
Non-immunological disorders
Severely retarded growth, significant clubbing, strabismus as a result of abducens nerve palsy along with hemiplegia, cerebral mass
Recurrent infections
Lymphoid interstitial pneumonia (LIP). Pleuropneumonia, chronic lung disease, bilateral bronchiectasis
Patient 2
Autoimmune
Self-limiting ITP; Asthma satisfactorily treated with inhaled steroids. Reactive monoarthritis responded well to local steroid injections
Immunologic investigations
Low immunoglobulin levels
Non-immunological disorders
Growth retarded (both weight and height were below the fifth percentile since the age of 2)
Recurrent infections
Serous otitis media, massive pneumonia along with loculated empyema, chronic lung disease with bilateral bronchiectasis
Patient 3
Chronic diarrhea
Severe diarrhea with no detectable bacterial or parasitic infection
Autoimmune
ITP, lymphadenopathy, autoimmune haemolytic anemia (AIHA), atrophic gastritis with autoantibodies against intrinsic factor, and a submaxillar abscess, granulomatous infiltration with T cells, plasma cells, and macrophages but showed low B cell numbers
Immunologic investigations
A moderate IgG hypogammaglobulinemia and complete IgA deficiency but a normal neutrophil count; intermittently elevated IgM; swelling of hilar and mediastinal lymph nodes with a mixed lymphoid follicular hyperplasia with the absence of the follicular mantle zone
Recurrent infections
Perinealmolluscumcontagiosum, recurrent warts, recurrent mild respiratory infections, severe recurrent pneumonias, including several interstitial pneumonias, a lymphoid interstitial pneumonia and bronchiectasis
Treatment
The interstitial lung disease was treated with methylprednisolone. Attempts to taper steroids were frequently associated with relapses, and long-term treatment with infliximab was initiated and allowed the discontinuation of methylprednisolone. Infliximab had little improvement of the chronic diarrhea. IVIG replacement
Patient 4
Chronic diarrhea and autoimmune
ITP, AIHA and autoimmune enteropathy (which can be classified as Crohn disease)
Non-immunological disorders
Failure to thrive
Immunologic investigations
Low immunoglobulin levels
Treatment
IVIG replacement and antibiotic prophylaxis
Recurrent infections
Recurrent upper-respiratory-tract infections, several episodes of pneumonia recurrent respiratory and gastrointestinal infections,severe lower respiratory- tract infections along with finger clubbing, hepatosplenomegaly an obstruction of the small airways and bronchiectasis, recurrent conjunctivitis and urticaria and a corpulmonale with consecutive right-heart failure
Treatment
Antibacterials, antifungals, and IVIG
Patient 5
Chronic diarrhea and autoimmune
Recurrent chronic diarrhea, autoimmune phenomena, hypothyroidism, and or AIHA allergic dermatitis, intestinal inflammation and subtotal villous atrophy, autoimmune phenomena, hypothyroidism, and myasthenia gravis. no signs of ITP or AIHA, bronchiectasis; Died at the age of 19 after respiratory failure
Non-immunological disorders
Retarded growth
Immunologic investigations
Selective IgA and IgG2 deficiency, IgG and IgMlevels declined gradually
Recurrent infections
Recurrent upper-respiratory-tract infections, including sinusitis and otitis media, pneumonia
Patient 6-10 from the study9.
Patient 6
Chronic diarrhea and autoimmune
Non-bloody diarrhea; partial villous blunting with intraepithelial lymphocytic infiltration; autoimmune pancytopenia; EBV-associated lymphoproliferative disease with intense infiltration with B cells
Immunologic investigations
Normal serum IgG, IgA, IgM, and IgE levels and normal numbers of CD3+ T lymphocytes, CD4+ and CD8+ T-cell subsets, B cells, and natural killer (NK) cells. Normal T-cell proliferation to the mitogens PHA and ConA� and a normal increase in antibody titers after vaccination with tetanus toxoid, diphtheria, toxoid and Haemophilusinfluenzae capsular antigens
Recurrent infections
no history of recurrent infections
Treatment
Responded to intravenous immunoglobulin (IVIG) replacement therapy and a short course of prednisone
Patient 7
Chronic diarrhea and autoimmune
Nephrotic syndrome; mucous nonbloody stools; lymphocytic infiltration of the lamina propria; villous atrophy and marked inflammation
Treatment
Prednisone and azathioprine with poor compliance; monthly intramuscular vitamin B12 injections; human growth hormone replacement
Immunologic investigations
Normal serum IgG, IgA, IgM, and IgE levels and normal numbers of CD3+ T lymphocytes, CD4+ and CD8+ T-cell subsets, B lymphocytes, and NK cells, normal T-cell proliferation to the mitogens PHA and ConA and a normal increase in antibody titers after vaccination with pneumococcal vaccine
Non-immunological disorders
Clubbing; growth hormone deficiency
Recurrent infections
No history of recurrent infections
Anemia
Megaloblastic anemia
Patient 8
Chronic diarrhea and autoimmune
Nonmucous and nonbloody chronic diarrhea; mucosal inflammation with lymphocytic infiltration but no granulomas or ulcerations; recurrent arthritis in the large joints, mainly the knees with inflammation
Treatment
Chronic diarrhea improved on oral prednisone
Immunologic investigations
Low serum IgG and IgA levels and decreased B-cell numbers but normal numbers of T cells, T-cell subsets, and NK cells; markedly reduced T-cell proliferation in response to PHA and anti-CD3 mAb
Recurrent infections
Recurrent otitis media and pneumonia; bilateral bronchiectasis and finger clubbing
Patient 9
Chronic diarrhea and autoimmune
Chronic nonbloody and nonmucous diarrhea; duodenal villous atrophy; autoimmune thrombocytopenia and autoimmune hemolytic anemia both of which responded to treatment with steroids and rituximab
Immunologic investigations
Low serum IgG and IgA levels and decreased B-cell numbers but normal numbers of T cells, T-cell subsets, and NK cells; markedly reduced T-cell proliferation in response to PHA and anti-CD3 mAb
Recurrent infections
no history of recurrent infections
Patient 10
Chronic diarrhea
Chronic diarrhea with no blood or mucus
Immunologic investigations
Low serum IgG and IgA levels and normal numbers of T cells, T-cell subsets, B cells, and NK cells; markedly reduced T-cell proliferation in response to PHA and anti-CD3 mAb
Recurrent infections
no history of recurrent infectionsor autoimmune hematologic manifestations
Patient 11the study8
Chronic diarrhea and autoimmune
Chronic diarrhea associated with an autoimmune enteropathy, duodenal villous atrophy and large bowel lymphocytic infiltration; erythema nodosum, transient arthritis of both feet, and recurrent hemolytic anemia; extensive lung infiltration of a mixture of CD3+ T and CD20+ B cells
Immunologic investigations
Raised IgG levels, raised inflammatory markers, and a low number of natural killer cells; normal lymphocyte subsets, double-negative T cells, T-cell proliferation assays, IgA, IgM, tetanus vaccine responses, and a nitrobluetetrazolium test; lymphadenopathy, splenomegaly, neutropenia, and thrombocytopenia, antineutrophil antibodies; normal CD19+ B cells and IgG level, a new-onset antibody deficiency with absent vaccine responses
Treatment
Several courses of steroids, rituximab (with prophylactic immunoglobulin replacement), and mycophenolatemofetil
Non-immunological disorders
Growth failure
Recurrent infections
No significant history of infections except for a psoas abscess associated with chronic neutropenia; five years after initial presentation, after multiple courses of rituximab, developed recurrent infections, after withdrawal of immunoglobulin therapy
Table 4: Clinical features of LRBA deficient patents.
Lopez-Herrera et al. reported on five subjects with LRBA mutations from four unrelated consanguineous families with childhood onset of CVID and autoimmunity [4]. Alangari A. et al. reported on five subjects with chronic inflammatory bowel diseases and CVID in a consanguineous family with deletions of two nucleotides in the LRBA gene [9]. Burns S et al. reported one female subject with autoimmunity and no hypogammaglobulinemia who had a large homozygous deletion that resulted in the absence of the LRBA gene [8]. Therefore, six LRBA germ line mutations cause CVID and autoimmunity with early onset (average 3 years of age) of symptoms [4,8,9], whereas the mean onset of CVID is 26.3 years of age [40]. Thus, the manifestations of LRBA deficiency include hypogammaglobulinemia due to defective B-cell differentiation, recurrent infections, particularly of the respiratory type, and various autoimmune disorders which include idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and inflammatory bowel diseases [4,8,10,18]. A subject died of respiratory failure at the age of 19 [4]. Other medical conditions such as severe retarded growth and failure to thrive, growth hormone deficiency, asthma, monoarthritis, seizures, granulomatous infiltration, finger clubbing, hepatosplenomegaly, allergic dermatitis, and nephrotic syndrome are also observed in LRBA deficient patients [4]. The clinical features of LRBA deficiency are listed in Table 4.
Protein structure of LRBA
The molecular weight of LRBA predicted from the largest open reading frame (ORF) of LRBA cDNA is 319 KD and similar size of protein was detected by Western blot [4]. Interestingly, mutated LRBA proteins cannot be detected in LRBA deficient patients by Western blot [4,8,9]. LRBA is composed of multiple domains [Figure 1] : the Concanavalin A (ConA)-like lectin binding domain [41], VHS [42] [VPS (vacuolar protein sorting)-27, Hrs (hepatocyte growth factor-regulated tyrosine kinase substrate) domain and STAM (signal transducing adaptor molecule)], RII binding motifs and WBW super domain. The three-dimensional structure of the WDL-BEACH of LRBA is described [43]. First, ConA is a lectin, which binds carbohydrate, originally extracted from the jack-bean, Canavaliaensiformis. ConA-like lectin domain was predicted in LRBA and other four LRBA paralogues [41]. It was proposed to bind oligosaccharides associated with protein traffick and sorting [41]. Second, the VHS domain is considered to have a general membrane targeting/cargo recognition role in vesicular trafficking [44]. It may be involved in vesicular trafficking by binding to sorting receptors that move and transfer cargo between the trans-Golgi network and the endosomal compartment [45]. The VHS domain in LRBA was predicted through a remote and functional conserved domain prediction algorithm [42]. Third, LRBA contains two potential RII binding sites for anchoring PKA through the RII subunits [2,46]. Finally, the WBW super-domain at the C-terminal is composed of a WDL and BEACH domain and five WD40 repeats. This same superdomain C-terminal architecture is shared by various large proteins which defined the WBW family. The WDL is structurally similar to the pleckstrin homology (PH) domain and strongly interacts with the BEACH domains. The interface between WDL and BEACH two domains forms a prominent groove which may be used to recruit binding partners [43,47]. LRBA GFP fusion protein is associated with vesicles, suggesting that BEACH and/or WD40 domains are involved in vesicle-binding or can form a dimer with LRBA or other proteins including WBW proteins [2].
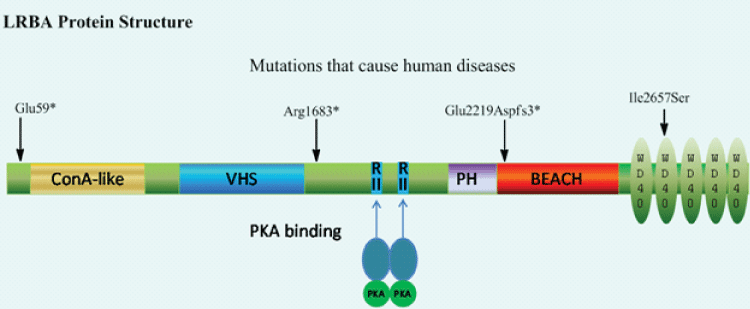
Figure 1: LRBA protein structure. Human LRBA protein has 2863 amino acids composed of multiple domains. The predicted molecular weight is 319 kD. ConA-like: Concanavalin A (ConA)-like lectin binding domain; VHS: VPS (vacuolar protein sorting)-27, Hrs (hepatocyte growth factor-regulated tyrosine kinase substrate) domain and STAM (signal transducing adaptor molecule); RII: PKA regulatory subunit (RII) binding motifs; PH: Plekstrin Homology (PH)-like domain; BEACH: beige and CHS domain; *= stop codon; fs*3=3 frame-shift codons then stop codon.
Deficiency of LRBA suppresses autophagy by about 50%, however, the molecular mechanism by which it does so is unknown. Our laboratory has identified a potential microtubule-associated protein 1 light chain 3 (LC3) interaction region(LIR) motif in both human and murine LRBA. The LIR consensus sequence is [DE]-[DE]-[DE]- [WFY]-X-X-[LIV] [48]. The DDDYVEL sequence from LRBA has high homology with LIRs from TP53INP1 (similarity is 92%) and TP53INP2 [49] (Table 1). Three dimensional structure remodeling (YASARA v12.7.16) shows that the LIR forms a complex with LC3 which is more stable than with p62, a known LC3 binding protein (Figure 2A). Tyrosine phosphorylation in the LIR destabilizes this complex (Figure 2B). The interaction energy between LC3 and LIRs of LRBA and three known LC3 binding proteins, the phosphorylated LIR of LRBA and the alanine scan were calculated using Foldx [50] (Table 3). LRBA may serve as an adaptor for ubiquitinized proteins destined for degradation through an association with LC3 and ubiquitin by LIR and an ubiquitin binding domain (UBD), respectively, during autophagy. Tyrosine phosphorylation may impede LRBA binding with LC3, inhibit autophagy, but favor cell growth, giving credence to the fact that LIR is phosphorylated in several forms of cancer cell lines such as lung, breast, bladder, kidney, gastric cancers and leukemia (https://www.phosphosite.org). These results may provide a molecular mechanism for clinical intervention of autophagy and cell growth related to human diseases.
![]()
LRBA-LIR
p62-LIR
LRBA-LIR -p
TP53INP1-LIR
TP53INP2-LIR
Alanine Scan
Interaction energy with LC3 (kcal/mol)
-15.19
-13.89
-13.22
-18.35
-15.13
-3.76
Interaction
++
+
+
+++
++
-
predicted
experimental
predicted
experimental
experimental
predicted
Table 3: Calculated interaction energy between LC3 and LIRs of other proteins using Foldx.
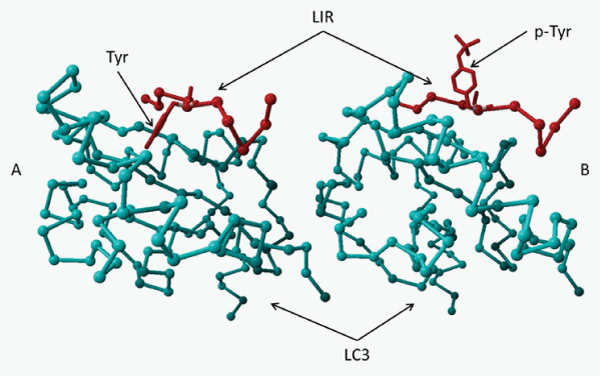
Figure 2: Three dimensional structure remodeling of LIR and LIC3 interaction A. The LIR (Red) from LRBA is predicted to form a complex with LC3 (Aqua) more stable than that of p62, a known LC3 binding protein. Tyrosine is in a hydrophobic pocket. B. Phosphorylation of the tyrosine in the LIR destabilizes the complex. Phospho-Tyrosine is hydrophibic. Its side chain is away from the hydrophobic pocket and the LC3.
In summary, LRBA potentially may serve as a scaffold to interact with multiple proteins involved in the endomembrane system/vesicle trafficking. However, the function of these domains has yet to be determined [37].
Paralogues of LRBA gene and disease formation
The exact molecular mechanism of LRBA remains unknown [37]. More valuable information about this gene may be obtained from the studies on its homologues, because structural similarities suggest functional similarities and vice versa. Bioinformatics analyses show LRBA is conserved in organisms ranging from unicellular to multicellular and probably exists in the whole eukaryotic kingdom and in all cell types. LRBA belongs to the WBW gene family containing the WBW super domain. WBW proteins appear to function as scaffolding proteins in vesicle trafficking [2,4,7,37] and are important in human diseases [37]. There are eight human WBW proteins: LRBA, lysosomal trafficking regulator (LYST), neutral sphingomyelinase activation associated factor (NSMAF), WD and FYVE zinc finger domain containing protein 3 (WDFY3), WD and FYVE zinc finger domain containing protein 4 (WDFY4), neurobeachin-like 1 (NBEAL1), neurobeachin-like 2 (NBEAL2), neurobeachin (NBEA), WD repeat domain 81 (WDR81) [37]. They appear to be involved in vesicle trafficking as scaffolding proteins and are involved inregulating lysosome size (LYST and NSMAF), apoptosis (NSMAF, LRBA), autophagy (LYST, WD and WDFY3, LRBA), granule size (LYST, NBEAL2, and NBEA), or synapse formation (NBEA) [2,4,7,37].
These family members are important in human diseases [37]. For example, mutations of four WBW genes (LYST, LRBA, NBEAL2 and WDFY4) cause recessive Mendelian diseases and NBEA is a currentcandidate for autism [37]. Similar to LRBA, LYST mutations cause Chediak-Higashi syndrome (CHS), a lethal human disorder with severe immunodeficiency and multiple organ lymphocyte infiltrations [4,8-10,51-53]. The platelets in CHS patients lack dense granules [54], while the platelets in the patients with gray platelet syndrome (GPS) caused by NBEAL2 mutations, lack alpha granules containing several growth factors [37]. WDFY4 is strongly associated with SLE in Asian populations [37]. NSMAF is a TNF adaptor protein which is required for the TNF-induced expression of cytokines, such as IL-6 and CXCL-2 [55,56]. LYST is required for LPS induction of inflammatory cytokines [57]. The Lyst deficient cells exhibit defective TLR signaling, specifically in the TLR4 pathways [57]. TLR4/LPS complex rapidly cycles between the cell membrane, the GC and endosomes [58,59], and can activate signaling pathways at the endolysosome [57], indicating vesicle-mediated localization and trafficking of TLR4 are required for its activation. These data suggest that LRBA may regulate cytokines and TLR4. Recurrent bacterial infections in LRBA-deficient patients may indicate an impaired TLR4/LPS pathway.
NBEA, which has 75% of protein homology with LRBA, is an isoform of LRBA and an autism candidate gene. It has very similar sub cellular localizations to that of LRBA and is implicated in post- Golgi membrane traffic and the regulatory secretion pathway of large dense-core vesicles containing growth factors and hormones [7,60-62]. NBEA binds to an important signaling complex, PKA. Interestingly, NBEA is a negative regulator of vesicle secretion as is the Beige Protein Homolog 1 (Bph1) in yeast. NBEA is related to the body length of mice as well as the synaptic spine patterns of neurons in the same animal; autism, platelet development and multiple myeloma in humans; and obesity in both mice and humans [37]. NBEA homozygous knockout mice died perinatally due to the lack of synaptic transmissions [63,64]. Deletion of NBEA reduces synaptic numbers and changes the localization of actin filaments in cultured neurons. This suggests that it may be important in trafficking cargo proteins to pre- and post-synaptic compartments [65]. NBEA is also a candidate tumor suppressor gene in multiple myeloma [66]. It also is involved in the regulatory secretion pathway of large dense-core vesicles containing growth factors and hormones [7,60-62].
Orthologue of the LRBA gene
LRBA orthologue is present in yeast as Bph1and in Drosophila as rugose (rg). Deletion of Bph1 causes increased secretion of carboxypeptidase Y in Saccharomyces cerevisiae and missorting of alkaline phosphatase. Bph1 is not essential and is both cytosolic and membrane peripherally bound. The vacuole morphology is not affected by disruption or overexpression of Bph1. The growth of the delta Bph1 strain is impaired by low pH, potassium acetate or calcofluor white, a fluorescent stain that binds strongly to the cell wall which contains cellulose and chitin, probably due to a defect on trafficking from the GC. Genetically, Bph1 interacts with VPS9, FLO1, FLO9, BTS1, OKP1, VPS9, BTS1 and OKP1. These data suggest that Bph1is involved in protein sorting and cell wall formation [67].
LRBA orthologue in Dictyostelium is essential for cytokinesis [68]. LRBA orthologue, sel-2, also called F10F2.1, in Caenorhabditiselegans, is a negative regulator of lin-12/Notch activity in the vulval precursor cells, which are polarized epithelial cells maintained through regulated activity of the basolateral LET-23/EGF receptor and apical LIN-12/Notch. Loss of sel-2 activity causes basolateral mislocalization and increased accumulation of LIN-12/Notch and basolateral LET- 23/EGF, indicating that SEL-2 is involved in endosomal traffic and may be involved in the efficient delivery of cell surface proteins to the lysosomes [69]. Two RII binding motifs were predicted for sel-2, both human and murine LRBA, by aligning with the known B1 and B2 PKA RII tethering sites in rg [2].
Mutations of rg in Drosophila are embryonic semi-lethal. These flies have a shorter lifespan and severe rough eye phenotype, caused by cell type-specific apoptosis with increased Jun N-terminal kinase activity and decreased EGFR signaling activity [70]. Genetically, rg interacts with 14 genes, including multiple components of EGFR, Notch, RAS and MAPK pathways [71,72].
Conclusion
LRBA is a novel gene essential for the normal function of the immune system and is significantly different from seven other CVID genes. It may regulate the following CVID genes: CD19, CD20 and BAFFR, because their levels are lower when LRBA is absent [4]. It encodes a large multiple domain protein, which may serve as a scaffold to interact with multiple proteins including other membrane regulators and cargo proteins. Vesicle trafficking is essential for homeostasis (plasma membrane deposition, recycling and degradation) and activation (oligomerization, phosphorylation and internalization) of cell membrane receptors, cytokine secretion and other proteins essential for normal function of the immune system. Although LRBA is extensively associated with the endomembrane system, including the GC, lysosomes, ER, plasma membrane, and perinuclear ER or nucleus, it may not be a constitutive structural component of these organelles, but rather a regulator of these structures, especially vesicle trafficking of cargo proteins between these membrane compartments. The cargo proteins may include cytokines and immunoglobulins for secretion, plasma membrane proteins for disposition on the membrane, and proteins trafficking between different membrane compartments, or proteins for degradation through lysosome/autophagy or proteasome degradation. Specifically, it interacts with multiple important signal transduction pathways, including EGFR, Notch, PKA, Ras, E2F1, p53 and MAPK [7,69,71,72]. These molecular interactions may help to understand why and how LRBA is involved in cell proliferation, apoptosis and autophagy [4,7] and plays a fundamental role in the normal immune system.
Acknowledgments
This work was supported by the Joy McCann Culverhouse Endowment to the Division of Allergy and Immunology, Department of Internal Medicine. We would like to thank Mrs. Geeta Gehi for helping preparing this manuscript.
References
- Kerr WG, Heller M, Herzenberg LA. Analysis of lipopolysaccharide-response genes in B-lineage cells demonstrates that they can have differentiation stage-restricted expression and contain SH2 domains. Proc Natl Acad Sci USA. 1996; 93: 3947-3952.
- Wang JW, Howson J, Haller E, Kerr WG. Identification of a novel lipopolysaccharide-inducible gene with key features of both A kinase anchor proteins and chs1/beige proteins. J Immunol. 2001; 166: 4586-4595.
- Feuchter AE, Freeman JD, Mager DL. Strategy for detecting cellular transcripts promoted by human endogenous long terminal repeats: identification of a novel gene (CDC4L) with homology to yeast CDC4. Genomics. 1992; 13: 1237-1246.
- Lopez-Herrera G, Tampella G,` Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012; 90: 986-1001.
- Kristensen AR, Gsponer J, Foster LJ. A high-throughput approach for measuring temporal changes in the interactome. Nat Methods. 2012; 9: 907-909.
- Kholodenko BN. Four-dimensional organization of protein kinase signaling cascades: the roles of diffusion, endocytosis and molecular motors. J Exp Biol. 2003; 206: 2073-2082.
- Wang JW, Gamsby JJ, Highfill SL, Mora LB, Bloom GC, et al. Deregulated expression of LRBA facilitates cancer cell growth. Oncogene. 2004; 23: 4089-4097.
- Burns SO, Zenner HL, Plagnol V, Curtis J, Mok K . LRBA gene deletion in a patient presenting with autoimmunity without hypogammaglobulinemia. J Allergy Clin Immunol. 2012; 130: 1428-1432.
- Alangari A, Alsultan A, Adly N, Massaad MJ, Kiani IS . LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol. 2012; 130: 481-488.
- Alangari A, Alsultan A, Adly N, Massaad MJ, Kiani IS. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol. 2012; 130: 481-488.
- Spickett GP, Farrant J, North ME, Zhang JG, Morgan L . Common variable immunodeficiency: how many diseases? Immunol Today. 1997; 18: 325-328.
- Saiki O, Ralph P, Cunningham-Rundles C, Good RA . Three distinct stages of B-cell defects in common varied immunodeficiency. Proc Natl Acad Sci U S A. 1982; 79: 6008-6012.
- Bryant A, Calver NC, Toubi E, Webster AD, Farrant J . Classification of patients with common variable immunodeficiency by B cell secretion of IgM and IgG in response to anti-IgM and interleukin-2. Clin Immunol Immunopathol. 1990; 56: 239-248.
- Gathmann B, Binder N, Ehl S, Kindle G, ESID Registry Working Party. The European internet-based patient and research database for primary immunodeficiencies: update 2011. Clin Exp Immunol. 2012; 167: 479-491.
- Eibel H, Salzer U, Warnatz K. Common variable immunodeficiency at the end of a prospering decade: towards novel gene defects and beyond. Curr Opin Allergy Clin Immunol. 2010; 10: 526-533.
- Park MA, Li JT, Hagan JB, Maddox DE, Abraham RS. Common variable immunodeficiency: a new look at an old disease. Lancet. 2008; 372: 489-502.
- Podjasek JC, Abraham RS. Autoimmune cytopenias in common variable immunodeficiency. Front Immunol. 2012; 3: 189.
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999; 92: 34-48.
- van Zelm MC, Reisli I, van der Burg M, Castaño D, van Noesel CJ. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006; 354: 1901-1912.
- Thiel J, Kimmig L, Salzer U, Grudzien M, Lebrecht D. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J Allergy Clin Immunol. 2012; 129: 801-810.
- van Zelm MC, Smet J, Adams B, Mascart F, Schandené L . CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010; 120: 1265-1274.
- Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IA. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest. 2010; 120: 214-222.
- Salzer U, Chapel HM, Webster AD, Pan-Hammarström Q, Schmitt-Graeff A. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005; 37: 820-828.
- Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A. 2009; 106: 13945-13950.
- Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003; 4: 261-268.
- Yong PF, Salzer U, Grimbacher B. The role of costimulation in antibody deficiencies: ICOS and common variable immunodeficiency. Immunol Rev. 2009; 229: 101-113.
- Fearon DT, Carroll MC. Regulation of B lymphocyte responses to foreign and self-antigens by the CD19/CD21 complex. Annu Rev Immunol. 2000; 18: 393-422.
- Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol. 2013; 131: 959-971.
- Thompson JS, Bixler SA, Qian F, Vora K, Scott ML. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science. 2001; 293: 2108-2111.
- Shulga-Morskaya S, Dobles M, Walsh ME, Ng LG, MacKay F . B cell-activating factor belonging to the TNF family acts through separate receptors to support B cell survival and T cell-independent antibody formation. J Immunol. 2004; 173: 2331-2341.
- von Bülow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity. 2001; 14: 573-582.
- He B, Santamaria R, Xu W, Cols M, Chen K. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol. 2010; 11: 836-845.
- Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005; 37: 829-834.
- Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity. 2003; 18: 279-288.
- Bartok B, Silverman GJ. Development of anti-CD20 therapy for multiple sclerosis. Exp Cell Res. 2011; 317: 1312-1318.
- Lamore R 3rd, Parmar S, Patel K, Hilas O. Belimumab (benlysta): a breakthrough therapy for systemic lupus erythematosus. P T. 2012; 37: 212-226.
- Cullinane AR, Schäffer AA, Huizing M. The BEACH is hot: a LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic. 2013; 14: 749-766.
- Parvaneh N, Casanova JL, Notarangelo LD, Conley ME. Primary immunodeficiencies: a rapidly evolving story. J Allergy Clin Immunol. 2013; 131: 314-323.
- Salzer U, Bacchelli C, Buckridge S, Pan-Hammarström Q, Jennings S. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009; 113: 1967-1976.
- Chapel H, Lucas M, Lee M, Bjorkander J, Webster D. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008; 112: 277-286.
- Burgess A, Mornon JP, de Saint-Basile G, Callebaut I. A concanavalin A-like lectin domain in the CHS1/LYST protein, shared by members of the BEACH family. Bioinformatics. 2009; 25: 1219-1222.
- Bradshaw CR, Surendranath V, Henschel R, Mueller MS, Habermann BH. HMMerThread: detecting remote, functional conserved domains in entire genomes by combining relaxed sequence-database searches with fold recognition. PLoS One. 2011; 6: e17568.
- Gebauer D, Li J, Jogl G, Shen Y, Myszka DG. Crystal structure of the PH-BEACH domains of human LRBA/BGL. Biochemistry. 2004; 43: 14873-14880.
- Misra S, Beach BM, Hurley JH. Structure of the VHS domain of human Tom1 (target of myb 1): insights into interactions with proteins and membranes. Biochemistry. 2000; 39: 11282-11290.
- Lohi O, Poussu A, Mao Y, Quiocho F, Lehto VP. VHS domain -- a longshoreman of vesicle lines. FEBS Lett. 2002; 513: 19-23.
- Hou T, Li Y, Wang W. Prediction of peptides binding to the PKA RIIalpha subunit using a hierarchical strategy. Bioinformatics. 2011; 27: 1814-1821.
- Jogl G, Shen Y, Gebauer D, Li J, Wiegmann K. Crystal structure of the BEACH domain reveals an unusual fold and extensive association with a novel PH domain. EMBO J. 2002; 21: 4785-4795.
- Birgisdottir ÅB, Lamark T, Johansen T . The LIR motif - crucial for selective autophagy. J Cell Sci. 2013; 126: 3237-3247.
- Seillier M, Peuget S, Gayet O, Gauthier C, N'Guessan P. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ. 2012; 19: 1525-1535.
- Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F. The FoldX web server: an online force field. Nucleic Acids Res. 2005; 33: W382-388.
- Faigle W, Raposo G, Tenza D, Pinet V, Vogt AB. Deficient peptide loading and MHC class II endosomal sorting in a human genetic immunodeficiency disease: the Chediak-Higashi syndrome. J Cell Biol. 1998; 141: 1121-1134.
- Barrat FJ, Le Deist F, Benkerrou M, Bousso P, Feldmann J. Defective CTLA-4 cycling pathway in Chediak-Higashi syndrome: a possible mechanism for deregulation of T lymphocyte activation. Proc Natl Acad Sci U S A. 1999; 96: 8645-8650.
- Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol. 2008; 15: 22-29.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab. 1999; 68: 283-303.
- Adam-Klages S, Adam D, Wiegmann K, Struve S, Kolanus W. FAN, a novel WD-repeat protein, couples the p55 TNF-receptor to neutral sphingomyelinase. Cell. 1996; 86: 937-947.
- Montfort A, de Badts B, Douin-Echinard V, Martin PG, Iacovoni J. FAN stimulates TNF(alpha)-induced gene expression, leukocyte recruitment, and humoral response. J Immunol. 2009; 183: 5369-5378.
- Cheng, W, Baier A, Lee K. Novel functions of a regulator of lysosomal trafficking, LYST in TLR4 signal transduction. Immunology. 2012; 137: 299-300.
- Latz E, Visintin A, Lien E, Fitzgerald KA, Monks BG. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J Biol Chem. 2002; 277: 47834-47843.
- Thieblemont N, Wright SD. Transport of bacterial lipopolysaccharide to the golgi apparatus. J Exp Med. 1999; 190: 523-534.
- Volders K, Scholz S, Slabbaert JR, Nagel AC, Verstreken P. Drosophila rugose is a functional homolog of mammalian Neurobeachin and affects synaptic architecture, brain morphology, and associative learning. J Neurosci. 2012; 32: 15193-15204.
- Castermans D, Volders K, Crepel A, Backx L, De Vos R. SCAMP5, NBEA and AMISYN: three candidate genes for autism involved in secretion of large dense-core vesicles. Hum Mol Genet. 2010; 19: 1368-1378.
- Wang X, Herberg FW, Laue MM, Wullner C, Hu B. Neurobeachin: A protein kinase A-anchoring, beige/Chediak-higashi protein homolog implicated in neuronal membrane traffic. J Neurosci. 2000; 20: 8551-8565.
- Su Y, Balice-Gordon RJ, Hess DM, Landsman DS, Minarcik J. Neurobeachin is essential for neuromuscular synaptic transmission. J Neurosci. 2004; 24: 3627-3636.
- Medrihan L, Rohlmann A, Fairless R, Andrae J, Döring M. Neurobeachin, a protein implicated in membrane protein traffic and autism, is required for the formation and functioning of central synapses. J Physiol. 2009; 587: 5095-5106.
- Niesmann K, Breuer D, Brockhaus J, Born G, Wolff I. Dendritic spine formation and synaptic function require neurobeachin. Nat Commun. 2011; 2: 557.
- O'Neal J, Gao F, Hassan A, Monahan R, Barrios S. Neurobeachin (NBEA) is a target of recurrent interstitial deletions at 13q13 in patients with MGUS and multiple myeloma. Exp Hematol. 2009; 37: 234-244.
- Shiflett SL, Vaughn MB, Huynh D, Kaplan J, Ward DM. Bph1p, the Saccharomyces cerevisiae homologue of CHS1/beige, functions in cell wall formation and protein sorting. Traffic. 2004; 5: 700-710.
- Kwak E, Gerald N, Larochelle DA, Vithalani KK, Niswonger ML. LvsA, a protein related to the mouse beige protein, is required for cytokinesis in Dictyostelium. Mol Biol Cell. 1999; 10: 4429-4439.
- de Souza N, Vallier LG, Fares H, Greenwald I. SEL-2, the C. elegans neurobeachin/LRBA homolog, is a negative regulator of lin-12/Notch activity and affects endosomal traffic in polarized epithelial cells. Development. 2007; 134: 691-702.
- Wech I, Nagel AC. Mutations in rugose promote cell type-specific apoptosis in the Drosophila eye. Cell Death Differ. 2005; 12: 145-152.
- Schreiber SL, Preiss A, Nagel AC, Wech I, Maier D. Genetic screen for modifiers of the rough eye phenotype resulting from overexpression of the Notch antagonist hairless in Drosophila. Genesis. 2002; 33: 141-152.
- Shamloula HK, Mbogho MP, Pimentel AC, Chrzanowska-Lightowlers ZM, Hyatt V. rugose (rg), a Drosophila A kinase anchor protein, is required for retinal pattern formation and interacts genetically with multiple signaling pathways. Genetics. 2002; 161: 693-710.
- Yan M, Wang H, Chan B, Roose-Girma M, Erickson S. Activation and accumulation of B cells in TACI-deficient mice. Nat Immunol. 2001; 2: 638-643.
- Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 1999; 18: 6910-6924.
- Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000; 404: 995-999.
- Wehr C, Eibel H, Masilamani M, Illges H, Schlesier M. A new CD21low B cell population in the peripheral blood of patients with SLE. Clin Immunol. 2004; 113: 161-171.
- Warnatz K, Wehr C, Dräger R, Schmidt S, Eibel H. Expansion of CD19(hi)CD21(lo/neg) B cells in common variable immunodeficiency (CVID) patients with autoimmune cytopenia. Immunobiology. 2002; 206: 502-513.