
Short Communication
Austin J Clin Immunol. 2014;1(1): 1005.
CD39 May Impact Clopidrogrel Pharmacodynamics
Joanna Lecka1,2, Michel Fausther1,2, Mireia Martín-Satué1,2,3 and Jean Sévigny1,2*
1Département de microbiologie-infectiologie et d’immunologie, Faculté de Médecine, Université Laval, Québec, QC, Canada.
2Département of immunologie,Centre de recherche du CHU de Québec, Québec, QC, Canada.
3Departament Patologiai Terapèutica Experimental, Facultat de Medicina, Universitat de Barcelona-Institut d’InvestigaticióBiomèdica de Bellvitge, Barcelona, Spain
*Corresponding author: Jean Sévigny, Centre de recherche du CHU de Québec, 2705, Boulevard Laurier, local T1-49, Québec, QC, G1V 4G2, Canada
Received: January 09, 2014; Accepted: January 14 , 2014; Published: January 17, 2014
Keywords
NTPDase1; clopidogrel; pro-drug; thrombosis; PON1; P2Y12
Thrombosis is treated with antithrombotic drugs such as clopidogrel. Clopidogrel which is widely prescribed after heart attacks is a pro-drug that must be metabolically activated to the form that irreversibly blocks platelet P2Y12 receptors. Bouman et al. [1] lately showed that the esterase paraoxonase-1 (PON1) was an enzyme involved in the catabolism of clopidogrel. Importantly, they showed that a common PON1gene polymorphismentails variability inclopidogrel’s clinical efficacy, a factor thought to be a major drawback to its use. This implies that PON1 genotyping might identify subjects unlikely to benefit from clopidogrel treatment. In agreement, E.Dolgin [2] described a case history of clopidogrel resistance correlating with low PON1 activity in a patient harbouring this genetic variant [3,4]. Taubert et al. [4] demonstrated that the P2Y12 antagonist is generated by the action of 2 enzymatic steps performed by cytochrome P450 (CYP2C19) and by PON1. They also showed that the latter is the ratelimiting step responsible for clopidogrel resistance. This explanation was confirmed by Homes MV et al. [5] in an extensive study, on over 42,000 patients, where no clinically significant interaction of CYP2C19 genotype with the association of clopidogrel therapy and cardiovascular events was shown.
The prognosis of patients on clopidogrel therapy that possess poor PON1 activity [1] might in fact be worse than expected. Indeed, Lecka et al. demonstrated that clopidogrel inhibits NTPDase1 activity [6]. NTPDase1 is responsible for ADP clearance from the blood and protects from uncontrolled platelet aggregation [7]. Indeed ADP is an important agonist of platelet activation and aggregation that is the natural ligand of P2Y12 and also of P2Y1, both expressed on platelets and responsible for their aggregation [7,8]. An oral dose of 30 mg clopidogrel, which constitutes 5% of the actual recommended therapeutic dosage, is expected to peak in the blood at ~20 μM. At this concentration, clopidogrel inhibited NTPDase1 activity, with either ATP or ADP as a substrate (>60% inhibition of ADPase activity). As a result, such inhibition favoured platelet aggregation in vitro [6]. These data are in agreement with the increased thrombosis and fibrin deposition found in cd39+/- mice in which the total enzyme activity of NTPDase1 (aka CD39) is reduced by half [7,9]. Conditions that diminish NTPDase1 activity have also been correlated with increased thrombosis and inflammation in several models [9-11].
Therefore, the previous comment suggesting that clopidogrel and its intermediary metabolites are “inactive” [12], on the assumption that they are not antagonists of P2Y12 receptors, may reveal to be wrong. The study by Leckaet al. [6] rather suggests that in the absence of the active metabolite of clopidogrel, the “P2Y12 inactive” clopidogrel pro-drug might in fact favour thrombosis by decreasing NTPDase1 activity [Figure 1]. This effect, which might be increased and/or prolonged in patients where the catabolism of clopidogrel to its active form is impaired, such as in subjects with low PON1 and/or CYP2C19 activity [2-4,12], would be expected to precede the therapeutic action of clopidogrel (i.e. dependent on drug metabolism). In agreement with this observation [6], we show here that 100 μM clopidogrel blocks the hydrolysis of ATP by endothelial NTPDase1 at the surface of both human umbilical vein endothelial cells (HUVEC) and also at the surface of blood vessels in liver and pancreas tissues ([Figure 2] and data not shown).
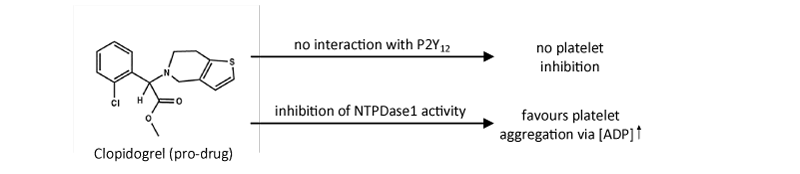
Figure 1: Schematic presentation of Topol and Schork’s comment as it appeared under “News & Views” [11] in relation with the article by Bouman et al. [1], shows that clopidogrel, in its pro-drug form, does not inhibit P2Y12 receptors and therefore does not block platelet aggregation (upper arrow). In a recent study [6] Lecka et al. showed that clopidogrel, at least in its pro-drug form, favours platelet aggregation via the build-up of ADP concentration at the endothelial cell surface due to the inhibition of NTPDase1 activity. Note that ADP is the natural ligand of P2Y1 and P2Y12 receptors and that by activating these receptors on platelet it plays a central role in regulating platelet function [7,8].
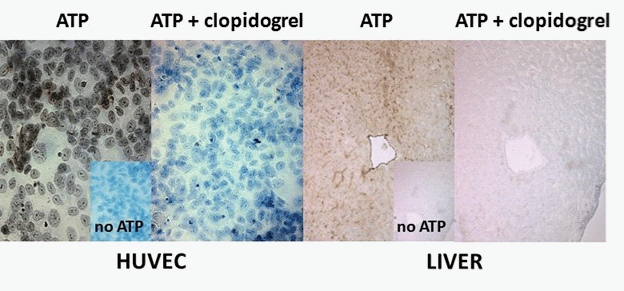
Figure 2: ATP hydrolysis at the surface of endothelial cells is blocked by clopidogrel. Freshly dissected thin sections of liver were embedded in O.C.T. freezing medium (Tissue-Tek®, Sakura Finetk, USA) and snap-frozen in isopentane in dry ice and stored at -80°C until use. Sections of 6 μm were prepared and fixed in 10% phosphate-buffered formalin mixed with cold acetone. Ectonucleotidase activity was located with the Wachstein-Meisel Pb(NO3)2 method [13]. Briefly, human liver sections or HUVEC grown on coverslips were fixed in 10% formalin in cold acetone, and further preincubated for 30 min at RT in 50 mM Tris-maleate buffer (pH 7.4) containing 2 mM CaCl2, 250 mM sucrose and 2.5 mM levamisole as an inhibitor of alkaline phosphatase. The enzymatic reaction was performed for 1 h at 37°C in the same buffer supplemented with 5 mM MnCl2, 2 mM Pb(NO3)2, 3% dextran T-250 ± 100 μM clopidogrel plus 200 μM ATP as substrate. For control experiments, the substrate was omitted. Reaction products were visualised by incubating the mixture with 1% (NH4)2S v/v for 1 min. These sections were counterstained with aqueous haematoxylin and mounted in Mowiol mounting medium and photographed under a BX51 Olympus microscope. Positive ATPase activity was detected at the surface of cultured HUVEC as well as in liver blood vessels. No reaction was found in the absence of ATP (no ATP, inset) or in the presence of clopidogrel. Scale bar, 50 nm.
In the light of this interpretation, patients who cannot appropriately convert clopidogrel to a P2Y12 antagonist, as in those exhibiting low PON1 and/or cytochrome 2C19 activity, might thus experience further complications from a therapy with this thienopyridine. Therefore, the addition of another antiplatelet drug together with clopidogrel, as is generally done, might be essential to ensure that NTPDase1 inhibition, albeit not yet tested in vivo, does not pose a threat for patientsduring clopidogrel therapy.
Acknowledgement
This work was supported by a joint grant from The Canadian Hypertension Society and Pfizer, and by grants from the Heart & Stroke Foundation (HSF) of Quebec and from the Canadian Institutes of Health Research (CIHR) to JS. JS was also a recipient of a senior Scholarship from the Fonds de recherche duQuébec � Santé (FRQS) and M. Martín-Satué of a fellowship from the Spanish Ministry of Education and Science (MEC-Programa José Castillejo). M. Fausther was the recipient of a Doctoral Scholarship from the Government of Gabon.
References
- Bouman HJ, Schömig E, van Werkum JW, Velder J, Hackeng CM, et al. Paraoxonase-1 is a major determinant of clopidogrelefficacy. Nat Med.2011; 17: 110-116.
- Dolgin E. Behind the paper: stepping up to the antiplatelet. Nat Med.2011; 17: 42.
- Taubert D, Bouman HJ, van Werkum JW. Cytochrome P-450 polymorphisms and response to clopidogrel. N Engl J Med.2009; 360: 2249-2250.
- vonBeckerath N, Taubert D, Pogatsa-Murray G, Schömig E, Kastrati A, et al. Absorption, metabolization, and antiplateleteffects of 300-, 600-, and 900-mg loading doses of clopidogrel: results of the ISAR-CHOICE (IntracoronaryStenting and AntithromboticRegimen: ChooseBetween 3 High Oral Doses for ImmediateClopidogrelEffect) Trial. Circulation.2005; 112: 2946-2950.
- Holmes MV, Perel P, Shah T, Hingorani AD, Casas JP. CYP2C19 genotype, clopidogrelmetabolism, plateletfunction, and cardiovascularevents: asystematicreview and meta-analysis. JAMA.2011; 306: 2704-2714.
- Lecka J, Rana MS, Sévigny J. Inhibition of vascularectonucleotidaseactivities by the pro-drugsticlopidine and clopidogrelfavoursplateletaggregation. Br J Pharmacol.2010; 161: 1150-1160.
- Enjyoji K, Sévigny J, Lin Y, Frenette PS, Christie PD, et al. Targeted disruption of cd39/ATP diphosphohydrolaseresults in disorderedhemostasis and thromboregulation. Nat Med.1999; 5: 1010-1017.
- Aslam M, Sedding D, Koshty A, Santoso S, Schulz R, et al. Nucleoside triphosphates inhibit ADP, collagen, and epinephrine-inducedplateletaggregation: role of P2Yâ‚ and P2Yâ‚ â‚ receptors. ThrombRes.2013; 132: 548-557.
- Guckelberger O, Sun XF, Sévigny J, Imai M, Kaczmarek E, et al. Beneficialeffects of CD39/ecto-nucleoside triphosphate diphosphohydrolase-1 in murine intestinal ischemia-reperfusioninjury. ThrombHaemost.2004; 91: 576-586.
- Lecka J, Gillerman I, Fausther M, Salem M, Munkonda MN, et al. 8-BuS-ATP derivatives as specific NTPDase1 inhibitors. Br J Pharmacol.2013; 169: 179-196.
- Chia JS, McRae JL, Thomas HE, Fynch S, ElkerboutL,et al. The protective effects of CD39 overexpression in multiple low-dose streptozotocin-induceddiabetes in mice. Diabetes. 2013; 62: 2026-2035.
- Topol EJ, Schork NJ Catapultingclopidogrelpharmacogenomicsforward. Nat Med.2011; 17: 40-41.
- Braun N, Sévigny J, Mishra SK, Robson SC, Barth SW,et al. Expression of the ecto-ATPase NTPDase2 in the germinal zones of the developing and adult rat brain. Eur J Neurosci.2003; 17: 1355-1364.