
School of Life Science, East China Normal University, China
*Corresponding author: Yuping Lai, Shanghai Key Laboratory of Regulatory Biology, School of Life Science, East China Normal University, 500 MinhangDongChuan Road, Shanghai, 200241, China
Received: August 15,, 2014; Accepted: November 14, 2014; Published: November 17, 2014
Citation: Wu Y, Li H, Jiang Z and Lai Y. The Interleukin-1 Family: A Key Regulator in the Pathogenesis of Psoriasis. Austin J Clin Immunol. 2014;1(5): 1023. ISSN : 2381-9138
Interleukin-1 (IL-1) family members, as most potent molecules of the innate immune system, are key regulators of multiple inflammatory diseases. The family includes seven ligands with agonist activity (IL-1α and IL-1β, IL-18, IL- 33, IL-36α, IL-36β, IL-36γ), three receptor antagonists (IL-1Ra, IL-36Ra, IL- 38), and one anti-inflammatory cytokine (IL-37). Most of these cytokines are abundantly expressed in skin under the regulation of IL-17 and act on innate immune cells to influence their survival and function. This review provides an overview of all the members of the IL-1 family and its potential regulatory roles in the pathogenesis of psoriasis.
Keywords: IL-1 family; IL-17; IL-17/IL-1 axis; Psoriasis
Interleukin-1(IL-1) family members, as the central mediator of innate immunity and inflammation, play a key role in the biology of multiple inflammatory diseases. So far, 11 members of the IL-1 family have been identified, including seven ligands with agonist activity (IL-1α and IL-1β, IL-18, IL-33, IL-36α, IL-36β, IL-36γ), three receptor antagonists (IL-1Ra, IL-36Ra, IL-38), and one anti-inflammatory cytokine (IL-37) (Table 1). According to the length of their precursor and the propiece for each precursor, the IL-1 family can also be categorized into three subfamilies including IL-1, IL-36 and IL-18. IL-1 family members signal through a group of closely related receptor complexes: IL-1R1 and IL-1RAcP complex as IL- 1α/β receptor, ST2 and IL-1RAcPcomplex as IL-33 receptor, IL-18Rα and IL-18Rβ complex as IL-18 receptor, and IL-1Rrp2 and IL-1RAcP complex as IL-36 receptor. The activation of these receptor complexes initiates and/or amplifies innate immune responses. Three other IL-1 receptor family members are IL-1R2, IL18BP and SIGIRR (also known as TIR8), which all act as negative regulators of IL-1 signaling [1,2].
Psoriasis is a common chronic inflammatory skin disease characterized by hyperplasia of epidermal keratinocytes and infiltrating immune cells. It is considered as a mixed Th1 and Th17 cell-mediated immune disease. Increasing evidence from experimental and clinical findings points to the important function of IL-1 family and IL-17 in the pathogenesis of psoriasis [3-8]. Most of IL-1 family members have been reported constitutively expressed by keratinocytes in vivo and shown to be highly expressed in the psoriatic skin [9-12]. This increasing expression of IL-1 family members contributes to Th17 cell development, leading to the production of IL-17 [13,14]. Therefore, IL-1 family is considered as an important mediator in the initiation and maintenance of psoriatic plaques. However, not only IL-1 family induces Th17 cells to produce IL-17, IL-17 in turn can act on keratinocytes to produce more IL-1 family cytokines [15,16]. These observations thereby indicate that the IL-17/IL-1 axis plays important roles in the pathogenesis of psoriasis. In this review, we summarize the experimental and clinical findings to consolidate our understanding on the function of IL-1 family members in psoriasis.
IL-1α and IL-1β: IL-1α (IL-1F1) and IL-1β (IL-1F2) are the first two members of IL-1 family discovered. Despite their identical activities, IL-1α and IL-1β have several differences. Firstly, IL-1β is secreted and circulates systemically, whereas IL-1α is generally associated with the plasma membrane of the producing cell and so acts locally. Secondly, IL-1β is mainly produced by monocytes and macrophages, whereas IL-1α is highly expressed by keratinocytes and endothelial cells. Thirdly, the two genes are differentially regulated during development and have different contributions during immune responses. Lastly, the pro-domain of IL-1α but not IL-1β has a nuclear localization sequence [17]. Although IL-1α and IL-1β have these differences, both of them bind to the same receptor complex including IL-1R1 and IL-1RAcP, and signal through myeloid differentiation primary response protein (MyD88). While this signaling can be negatively regulated by IL-1 receptor antagonist, IL- 1Ra (IL-1F3), which is the natural antagonist of IL-1α and IL-1β [18].
In skin lesions of psoriasis patients, IL-1β has been shown to be markedly increased, and effective treatment of psoriasis led to a significant decrease in epidermal IL-1β expression, suggesting that IL-1 subfamily plays a role in the pathogenesis of psoriasis [10,12]. Moreover, IL-17 was markedly increased in lesional skin of psoriasis patients, and these patients treated with Secukinumab, a fully human anti-IL-17A monoclonal antibody, had a reduction of 75% or more in the Psoriasis Area-and-Severity Index score (PASI75) [19-21]. However, it was not clear whether IL-17 regulated IL-1 expression in these studies. In 2013, the studies from Albanesi’s and Chen’s groups showed that IL-17A induced IL-1β expression in macrophages via the activation of MAPKs, NF-κB and AP-1[22], and IL-17, in combination of IL-4, markedly increased IL-1α and IL-1Ra in supernatants and cell lysates of cultured keratinocytes [16], while the studies from Cooper’s and Mee’s groups showed decreased IL-1α but increased IL-1β expression in lesional skin of psoriasis patients [23,24]. Moreover, when Muhr et al. compared IL-1 subfamily gene expression in keratinocytes from psoriasis patients and healthy individuals in the presence of IL-17, they found that IL-17 induced IL-1β but not IL-1α and IL-1Ra expression in keratinocytes from healthy individuals [15]. Therefore, how IL-17 specifically regulates the expression of IL-1 subfamily members in psoriasis needs further investigation.
To confirm the role of IL-1α, IL-1β and IL-1Ra in the pathogenesis of psoriasis in vivo, transgenic mice targeting related genes were generated. Skin lesions with hyper proliferative epidermis and increased antimicrobial transcripts including S100A7, S100A9, DEFB4 were observed in Il1a and Il1rn transgenic mice, suggesting IL-1α signaling enables to initiate an inflammatory reaction in psoriatic development [25,26]. In addition, the deficiency of IL-1Ra in mice resulted in spontaneous psoriatic-like lesions [27]. In human, children born with a genetic deficiency of Interleukin-1-Receptor Antagonist (IL-1RA) or functional inactive IL-1RA suffer from severe systemic and local inflammation, including pustular skin eruption [28,29]. Moreover, the profile of the transcriptome from psoriatic tissue and IL-1α-treated cultured human keratinocytes revealed a high correlation of the transcriptome profile between IL-1α-treated keratinocytes and psoriatic tissues, suggesting the inflammatory milieu in the epidermal microenvironment in psoriasis is more likely dependent on evolutionarily ancient cytokines such as IL-1α, rather than those of the adaptive immune response [30]. Furthermore, IL- 1β, as well as IFN-γ, was able to induce the psoriatic regenerative epidermal phenotype including keratin16, keratin17, and keratinocyte Transglutaminase (TGK), ICAM-1 and HLA-DR in both normal human skin and non-lesional skin from psoriatic patients, and IL- 1RA inhibited the effects of IFN-γ on the expression of keratin 17 and TGK in normal skin but not in non-lesional skin of patients with psoriasis, suggesting IL-1RA system may be dysregulated in psoriatic skin [31]. However, whether IL-1β acts directly or indirectly to these target genes remains unclear. Altogether, these findings demonstrate the tremendous impact of IL-1 subfamily members on psoriasis, and suggest that the blockage of IL-1 signaling might have a broader clinical impact on psoriasis.
IL-33(IL-1F11) is the most recently identified member of the IL-1 family and is a ligand for the orphan receptor ST2 [32]. In contrast to other IL-1 family members, it is not typically expressed by haematopoietic cells but is abundantly expressed in the endothelial cells and keratinocytes. In psoriasis patients, both IL-33 and ST2 significantly increased in lesional and non-lesional skin, compared to those in healthy skin [33-36]. The studies from Balato’s and Theoharides’s groups showed that IL-33 was strongly associated with endothelial cells in psoriatic skin compared to non-lesional psoriatic skin and healthy controls [33,36]. Moreover, IL-33 was secreted by psoriatic keratinocytes and was present in nucleus as well as cytoplasm in keratinocytes [33]. The expression of IL-33 was partially under the regulation of IL-17A. The study from Meephansan’s group showed that IL-17A induced IL-33 expression at mRNA and protein levels in time and dose-dependent manners in Normal Human Epidermal Keratinocytes (NHEKs). This induction was via the activation of EGFR, ERK, p38 and STAT3, as IL-17A-induced IL-33 expression was blocked by the addition of EGFR, ERK, p38 and STAT3 inhibitors [35]. However, an intrinsic mechanism by which IL-17 regulates IL- 33 expression in keratinocytes warrants further investigation.
IL-33, as a dual function protein, acts as both a cytokine to activate a number of immune cells with potent pro-inflammatory effects and intracellular nuclear factor to suppress pro-inflammatory gene transcription [37,38]. IL-33 induces mast cell degranulation, maturation and the production of IL-1, IL-6, IL-13, TNF-α, CCL2 and CCL3 in psoriatic lesional skin [39,40]. In 2010, a relationship between IL-33 and peptide Substance P (SP), VEGF in mast cell was well studied. IL-33 augmented the effect of SP on inducing mast cell release of VEGF, leading to increased angiogenesis in psoriasis [36]. Our previous data showed that IL-17 induced REG3A to regulate keratinocyte hyperproliferation in psoriasis [41], and the induction of REG3A by IL-17 was dependent on IL-33(unpublished data). These results thereby demonstrate the important role of IL-33 in psoriasis-like plaque formation and targeting IL-33 may provide a new treatment strategy for psoriasis.
IL-36 proteins: IL-36 proteins contain IL-36 ligands and IL- 36Ra. IL-36 ligands include IL-36α, IL-36β and IL-36γ (formerly IL- 1F6, IL-1F8 and IL-1F9) that signal through the IL-1 receptor family members such as IL-1Rrp2 (IL-1RL2) and IL-1RAcP [42-44]. IL- 36Ra (IL-1F5) is an antagonist of IL-36 signaling. IL-36Ra binds to IL-1Rrp2, blocks IL-36 ligand binding to the IL-1Rrp2 receptor and the subsequent recruitment of IL-1RAcP [45].
IL-36 is abundantly expressed in skin and a few other tissues. Their expression can be strongly induced in monocytes and keratinocytes [2]. Accumulating evidence suggests that IL-36 is a crucial cytokine in the pathogenesis of psoriasis [11,42,43,46]. In 2001, Debets et al. first reported that IL-36 proteins were over expressed in psoriatic lesional skin [43]. This observation was further confirmed by other scientists. Johnston’s group showed IL-36Ra (IL-1F5), IL-36α (IL- 1F6), IL-36β (IL-1F8), IL-36γ (IL-1F9) were significantly higher in psoriatic lesional skin than those in non-lesional skin [11]. Blumberg et al. also found increased expression of IL-1Rrp2, IL-36Ra and IL- 36α in human psoriatic skin [42]. Moreover, IL-36 has been shown to be under the regulation of IL-17. Carrier’s study reported that IL-36 cytokines were increased in a Th17-dominant psoriasis-like animal model [47]. Muhr’s study showed that IL-17 induced IL-1 family members IL-36α and IL-36γ, but not anti-inflammatory members IL- 1Ra, IL-36Ra and IL-37 in keratinocytes from psoriatic skin, when compared to those from healthy individuals [15].
To confirm the pathological role of IL-36 proteins in psoriasis development, IL-36α transgenic mouse was generated by Blumberg’s group. The mouse exhibited psoriatic skin phenotype with thickened scaly skin, acanthosis, hyperkeratosis and a mixed inflammatory cell infiltrate in the dermis [42]. Cytokines implicated in the pathogenesis of psoriasis, such as IL-23, IL-17 and TNF-α, were increased in this model, and they were induced in wild-type mouse skin by IL- 36α and in turn induced IL-36α production in keratinocytes. The combination of IL-36α transgene with an IL-36Ra deficiency resulted in the exacerbation of the psoriatic phenotype, demonstrating the antagonistic activity of IL-36Ra in vivo [42]. Moreover, the psoriatic phenotypes were ameliorated after IL-36α transgenic mice crossed with IL-36Ra transgenic mice. In summary, dysregulated expression of IL-36 agonists and antagonists promotes cutaneous inflammation, leading to psoriatic inflammatory skin disorders.
Increasing evidence further dissects the roles of IL-36 proteins in the pathogenesis of psoriasis. Towne’s group reported that IL-36α, IL- 36β and IL-36γ activated IL-8 promoter and induced IL-6 secretion through the MAPKs, JNK and ERK1/2 pathway [44]. Microarray analysis was performed by Johnston’s group after reconstituted epidermal cultures treated with recombinant IL-36α, IL-36β and IL-36γ. Strikingly, these cytokines not only induced IL-8 expression but also induced the expression of Antimicrobial Peptides (AMPs), Matrix Metalloproteinase’s (MMPs) and growth factors. In particular, IL-36β effectively induced HBD-2, HBD-3, MMP19 and MMP9 [11]. IL-36α and IL-36β but not IL-36γ directly induced TNF-α, IL-6, IL-8, hBD2, S100A7, and these effects of IL-36α and IL-36β were synergized with IL-17A and TNF-α [47]. In addition to keratinocytes, IL-36 subfamily has also been reported to target another cell types such as dendritic cells, as IL-36 receptor is expressed in this cell type [48-50]. In Bone Marrow-Derived Dendritic Cells (BMDCs), IL-36 induced the expression of IL-12, IL-1β, IL-6, TNF-α and IL-23. In addition, IL-36β enhanced the expression of CD80, CD86 and MHC class II by BMDCs [50]. Besides BMDCs, IL-36 induced the expression of IFN-γ, IL-4 and IL-17 in CD4+ T cells, suggesting a critical role of IL- 36 subfamily members in the stimulation of T helper cell responses [50].
Moreover, a link between IL-36Ra and Generalized Pustular Psoriasis (GPP), a different form of psoriasis, has been identified [51- 53]. Marrakchi et al. performed homozygosity mapping and direct sequencing in nine Tunisian multiplex families with autosomal recessive GPP and found that familial GPP was caused by the deficiency of IL-36Ra, that is, all the patients with GPP were found to carry a loss function mutation in IL-36RN. This aberrant IL-36Ra structure and function led to deregulated secretion of inflammatory cytokines (IL-1α, IL-6, IL-8, TNF-α) [51]. In 2013, Sugiura et al. reported that the majority of cases of GPP without a history of psoriasis vulgaris were caused by homozygous or compound heterozygous mutations of IL36RN, although only a few cases of GPP preceded or accompanied by psoriasis vulgar were found to have IL36RN mutations [52]. In 2014, a case of GPP was successful treated by Granulocyte and Monocyte Adsorption apheresis (GMA), suggesting that granulocytes/monocytes play a major role in the immunopathogenesis of GPP caused by deficiency of IL-36Ra [53].
IL-38(IL-F10) is originally identified in silicon. Its gene is located in the IL-1 family cluster on chromosome 2 next to the genes encoding IL-1R1 and IL-36Ra [1,54]. As one of three IL-1 family receptor antagonists, IL-38 shares 43% homology with IL-36Ra [54]. It binds to the IL-36R to inhibit IL-36 signaling as IL-36Ra does [55]. IL-38 polymorphism is associated with psoriatic arthritis, suggesting IL-38 might play a role in the pathogenesis of this inflammatory skin disease [56-58]. However, until now, no study reports that IL-38 is regulated by IL-17A. Conversely, addition of IL-38 in peripheral blood mononuclear cells inhibited the production of IL-22 and IL- 17A, suggesting IL-38 may be involved in the regulation of IL-17 expression [55].
IL-18(IL-1F4) and IL-37(IL-1F7) are expressed by macrophages and dendritic cells as well as epithelial cells, such as keratinocytes. Both IL-18 and IL-37 bind to the same receptor IL-18Rα. However, IL-18 acts as a proinflammatory factor, while IL-37 serves as a natural brake of inflammation. There is no direct evidence support that IL-17 induces the expression of IL-18 and IL-37, but it is reported that IL-18 was decreased in cuprizone-treated Act1 knockout mice compared to that in WT mice, indicating the essential role of IL-17-Act1-mediated signaling in the production of IL-18 [59]. More importantly, IL-18 has also been implicated in several autoimmune diseases such as psoriasis. Arican et al reported IL-18 expression was increased in the serum of psoriatic patients and correlated with disease severity [60]. However, so far no report shows that IL-37 is correlated with the pathogenesis of psoriasis.
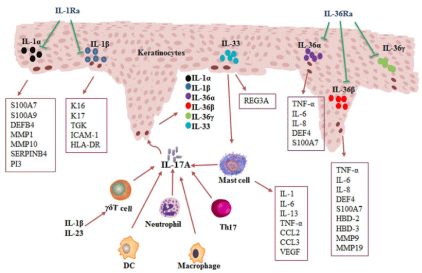
Psoriasis is a common immune-mediated inflammatory disease. The pathogenesis of psoriasis is a complex integration of genetic, immunological and environmental components [8]. IL-17/IL-1 axis exerts a proinflammatory effect on keratinocytes and immune cells in psoriasis. IL-17 secreted by Th17 cells and γδ T cells induces keratinocyte activation with the release of the proinflammatory cytokines such as IL-1 family members. IL-1, in turn, can act on T cells and induce Th17 cells differentiation from naïve precursors, as well as in the promotion of IL-17- and IL-22- producing T cells. These events sustain and amplify the chronic inflammation in psoriasis (Figure 1). Moreover, cytokines of IL-1 family induce the production of multiple antimicrobial peptides or proteins in keratinocytes, leading to hyperproliferation of skin epidermis. So far, biological agents that target IL-17 and its receptor have been developed for psoriasis treatment and shown promising effects on phase 3 clinical studies [19-21, 61-63]. However, although there is accumulating evidence supporting the involvement of IL-1 family cytokines in the pathogenesis of psoriasis and anti-IL-1 strategies have had a tremendous impact on the therapy of some auto-inflammatory disorders [18], scarce biological agent specifically targeting IL-1 is developed for the treatment of psoriasis. The expression of IL-1 family members, especially IL-36 subfamily members, is limited to skin, airway and a few other tissues, suggesting that its inhibition may have fewer systemic consequences. Therefore, one can speculate that IL-36 subfamily member might be a promising therapeutic target for the treatment of psoriasis. However, better understanding of the pathophysiology of IL-1 family cytokines in psoriasis is required to this end.
We apologize to those authors whose work could not be cited because of space constraints. Work in the laboratory of Yuping Lai is supported by National Natural Science Foundation of China grants 31222021, 31170867 & 31470878, grants 13JC1402301, 13SG25, 141017, 12ZZ039, NCET-11-0141 to Y.L, NSFC grant 81202327 to Yelin Wu, the program for professor of Special Appointment (Eastern Scholar) at Shanghai Institutions (Y.L) and the Science and Technology Commission of Shanghai Municipality grant 11DZ2260300.
Nomenclature and main functions of IL-1 family members in psoriasis.
Subfamily |
Family member |
Receptor(co-receptor) |
Expression in lesional skin of psoriasis |
Function in psoriasis |
Whether induced by IL-17 |
IL-1 subfamily |
IL-1α(IL-1F1) |
IL-1R1(IL-1RAcP) |
Low |
Proinflammatory |
Yes |
IL-1β(IL-1F2) |
IL-1R1(IL-1RAcP) |
High |
Proinflammatory |
Yes |
|
IL-1Ra(IL-1F3) |
IL-1R1 |
No change |
Antagonist for IL-1α, IL-1β |
Unknown |
|
IL-33(IL-1F11) |
ST2(IL-1RAcP) |
High |
Proinflammatory |
Yes |
|
IL-36 subfamily |
IL-36α(IL-1F6) |
IL-1Rrp2(IL-1RAcP) |
High |
Proinflammatory |
Yes |
IL-36β(IL-1F8) |
IL-1Rrp2(IL-1RAcP) |
High |
Proinflammatory |
Yes |
|
IL-36γ(IL1-F9) |
IL-1Rrp2(IL-1RAcP) |
High |
Proinflammatory |
Yes |
|
IL-36Ra(IL-1F5) |
IL-1Rrp2 |
High |
Antagonist for IL-36α, IL-36β, IL-36γ |
Unknown |
|
IL-38(IL-1F10) |
IL-1Rrp2 (IL-36R) |
High |
Antagonist for IL-36α, IL-36β, IL-36γ |
Unknown |
|
IL-18 subfamily |
IL-18(IL-1F4) |
IL-18Rα(IL-18Rβ) |
Unknown |
Unknown |
Unknown |
IL-37(IL-1F7) |
IL-18Rα |
Unknown |
Anti-inflammatory |
Unknown |
The pathological roles of IL-17-IL-1 axis in psoriasis.
Austin Publishing Group is an emerging open access publisher specialising in Science, Technology and Medicine is dedicated to serve the biomedical community through its initiatives. Austin Publishing Group is an academic publisher with 100+ peer reviewed open access journals in various subjects such as biomedical, Pharma, Life Sciences, Environmental, Engineering and Management. Austin Publishing Group publishes Open Access eBooks providing free access to vast scientific literature.