
Case Report
Austin J Clin Immunol. 2015;2(1):1024.
Orbital Pseudotumor - A Rare Variant of Granulomatosis with Polyangiitis
Majai G¹, Tarjan P², Dezso B³ and Zeher M¹*
¹Division of Clinical Immunology, University of Debrecen, Hungary
²Department of Internal Medicine, County Hospital Szolnok, Hungary
³Department of Pathology, University of Debrecen, Hungary
*Corresponding author: Margit Zeher, Division of Clinical Immunology, Faculty of Medicine, University of Debrecen, Móricz Zsigmond Street 22, Debrecen 4032, Hungary
Received: June 18, 2015; Accepted: June 25, 2015; Published: June 26, 2015
Abstract
Granulomatosis with Polyangiitis (GPA) is an autoimmune small vessel vasculitis with unknown aetiology, characterized by necrotizing granulomatous inflammation affecting the renal, pulmonary, upper airways and ocular systems and is often associated with circulating Anti-Neutrophil Cytoplasmic Antibodies (ANCA) against proteinase 3.
Here we report a presentation of GPA showing orbital involvement (without systemic involvement) with the classical histological triad, and also significant localized tissue eosinophilia which corresponds with the rare eosinophilic form of GPA. This variant of GPA should be differentiated from Eosinophilic Granulomatosis with Polyangiitis (EGPA); and the differential diagnosis between the two entities, and also the treatment of GPA is discussed.
Keywords: Orbital pseudotumor; Eosinophilic variants of GPA; Small vessel vasculitis; Anti-CD20 monoclonal antibody therapy
Abbreviations
GPA: Granulomatosis with Polyangiitis; EGPA: Eosinophilic Granulomatosis with Polyangiitis; ENT: Ear, Nose, Throat; ANCA: Anti-Neutrophil Cytoplasmic Antibodies; cANCA: cytoplasmic ANCA; MRI: Magnetic Resonance
Introduction
Granulomatosis with Polyangiitis (Wegener’s Granulomatosis, GPA) is an autoimmune small vessel vasculitis, characterized by necrotizing granulomatous inflammation of Ear, Nose, Throat (ENT), pulmonary, renal, ocular systems, and is often associated with circulating Anti-Neutrophil Cytoplasmic Antibodies (ANCA) against proteinase 3. Its clinical manifestation can be organ/life threatening. Two different phenotypes can be distinguished in GPA, namely the localized/limited and the systemic form [1].
The patients with limited disease have mostly ENT involvement including sinus involvement, nasal septal perforations, nasal collapse and subglottic stenosis. The presence of lung, renal, nervous system involvement is more frequent in patients with systemic form. It was shown that patients with limited disease are nearly a decade younger at disease onset, with longer disease duration compared to patients with severe disease, moreover, the female population is more frequent. The presence of ANCA occurs in 78% of the limited, and 90% of the systemic forms of GPA [2]. The ophthalmological manifestations are present at the diagnosis in about 34% of GPA patients [3] however a long term study showed that 52% of patients developed ophthalmologic manifestations during the course of the disease [4]. The most common ophthalmological manifestations are conjunctivitis (52.1%), episcleritis (39.3%), orbital inflammatory pseudo-tumour (19. 7%) and blurred vision (15.4%) [3].
Case Report
A 38-year-old woman was admitted to our hospital in April 2012 with unilateral exophthalmos, diplopia, and visual impairment. In April 2010 she was admitted to the neurology department with collapse, blurring vision and sweating. EEG, liquor examinations, routine laboratory test were performed and they were within normal range. The Magnetic Resonance Imaging (MRI) identified left side meningeal thickening. In October and November 2010 she presented oral aphtae and arthralgia. The routine laboratory test, hepatitis serology, chest radiography and abdominal ultrasound showed no infection or malignancy. The immunoserology was also negative. In June 2011 she was admitted to the ophthalmology with left side exophthalmos. The MRI identified left side meningeal thickening, left side mastoiditis, and left nonhomogenous orbital mass. At that time, the repeated autoantibody profile was negative. She was treated with high dose of corticosteroid and azathioprine. After 3 month of treatment a control MRI was done, the previously described orbital mass showed no changes. Thereafter an orbital biopsy was performed, and IgG4 level was determined to exclude IgG4 associated disease, malignancy, or orbital inflammation process. The steroid dose was reduced, after a while suspended, and she continued the azathioprin therapy only. During this time, the orbital mass began to increase and the left side exophthalmos became more pronounced. In January 2012 a new orbital biopsy was performed. Histological analysis revealed vasculitis, granulomas formation and tissue eosinophillia.
At her presentation in our department, a left upper eyelid hyperaemia, swelling and exophthalmos were observed. The overlying skin was mobile, the bony margins were palpable and draining lymph nodes were not enlarged. The ENT and systemic examination did not reveal any abnormality. She had no history of atopy, asthma, sinonasal, respiratory or renal disease.
Ophthalmological examination showed dislocation of the left eyeball, diplopia, myopia and secondary glaucoma. Hertel exophtalmometry was used to examine the ocular protrusion. The examination showed 16 mm on right side, 22mm on left side and the interorbital distance was 106mm.
The complete blood count, renal function, electrolytes, thyroid function test, anti –TPO antibody, urine examination did not revealed abnormality. The immunoserology, including ANCA was negative. The previously performed biopsy was requested by our clinic, and a new histological analysis was performed which showed fibroadipose tissue with inflammatory necrosis in the capillaries, venules and arterioles, associated with epitheloid granulomas formation and tissue eosinophillia (Figure 1).
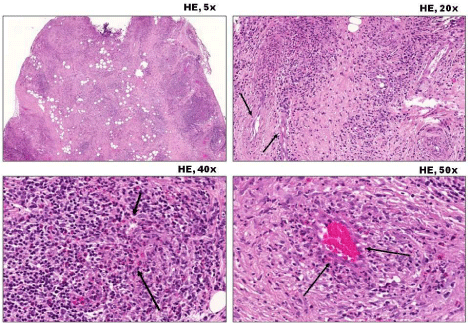
Figure 1: Hematoxylin-eosin stained images in various magnifications
(at upper right corner) from the orbital biopsy specimen are shown to
demonstrate granulomatous necrotizing vasculitis (arrows) with significant
tissue eosinophilia. Note the obvious granuloma on the right upper image with
vascular involvement (left hand side). Significant number of eosinophils can
easily be recognized on lower images. The histopathological investigation
revealed that 80% of lymphocytes were eosinophil granulocytes in the
perivascular infiltration.
The diagnosis of eosinophilic variant of GPA was made based on clinical history and histological findings. The patient was treated with oral methylprednisolone at dose of 1.5mg/kg with gradual tapering and monthly pulse cyclophosphamide, started at dose 600 mg/pulse. After six month of treatment, a control MRI was performed which showed no reduction of the orbital mass, but changes in the structure of the mass, and we started the rituximab medication 1000 mg in 2 weeks intervals. After 3 months, the local orbital oedema reduced, and on MRI result, a moderate regression was observed. Hertel examination was 16mm on right side, 18mm on left side and the interorbital distance was 106mm.
After six months, in September 2013 the second cycle of rituximab therapy was continued with the same protocol, the control MRI showed the same moderate regression of the orbital mass (Figure 2). According to the regression of clinical signs: disappearance of left upper eyelid hyperemia, swelling and diplopia and improvement in Hertel examination results (15mm protrusion on right side, 17mm on left side and the interorbital distance 102mm), the complete clinical regression was established. The therapy was continued with tapering dose of methylprednisolone and methotrexate.
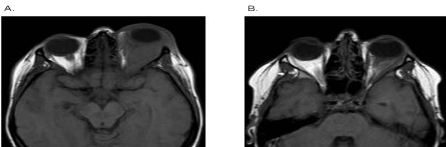
Figure 2A: Axial T1-weighted MRI imaging showing left side nonhomogenous
orbital pseudo tumor with retro bulbar adipose tissue oedema, but no
intracranial extension, before the treatment with rituximab. B. Axial T1-
weighted MRI scan showing left side orbital pseudo tumor regression after
the rituximab therapy.
Discussion
The diagnosis of GPA is based on the clinical manifestations, positive ANCA serology and histological characteristics of the disease.
The clinical symptoms of the disease often overlap with other inflammatory disorders. The diagnosis of localized ophthalmic GPA is difficult, because ANCA serology is positive in 32-78% only [2,5,6], therefore, biopsy for histology often should be performed.
The typical histological triad described in GPA is vasculitis, necrosis and granulomatous inflammation. It was shown that there are significant differences in the cellular profile in biopsies from patients with GPA. This biopsy showed more inflammatory cells compared to other orbital inflammatory disease. The presence of eosinophils is often exhibited in GPA (53%) [7], and this is suggested as a predictor of disease progression [8]. The eosinophilic variant of GPA is rare, and characterized by systemic /or localized disease with typical histological triad for GPA, but with additional tissue eosinophilia in the absence of asthma or atopy that would reflect typical Churg-Strauss syndrome [9-13].
It should be differentiated from Eosinophilic Granulomatosis with Polyangiitis (EGPA) which typically associated with tissue and systemic eosinophilia, clinical history of atopy and asthma. Peripheral eosinophilia is occasional only in GPA in contrast to the profound persistent tissue eosinophilia in patients with EGPA. In EGPA, IgE level is also increased in most of the cases due to some unrecognized extrinsic allergen. During the diagnosis, fungal, parasitic, bacterial infection should be also excluded.
After the diagnosis, treatment with high dose glucocorticoid in combination with another immunosuppressive therapy (cyclophosphamide, or rituximab) is the first line of treatment choice. In case of vital organ involvement, or in life threatening complication, combination with plasma exchange should be considered. Following the successful remission, cylophosphamide should be substituted with azathioprin or methotrexate for at least 24 months. Rituximab may be used as maintenance therapy, 1000 mg every 4-6 months for 2 years [14].
With our case, we would like to highlight that unilateral upper eyelid swelling and exophthalmos may be the first presentations of GPA. For appropriate diagnosis, careful investigations are important, even including a secondary biopsy. Positive ANCA may help in establishing the diagnosis, but in high percentage of patients with localized forms of the disease, cANCA may just as well be negative. The correct diagnosis of the disease and, in turn, aggressive treatment is important to prevent progressive disease manifestation with organ/ life threatening condition.
References
- Comarmond C, Cacoub P. Granulomatosis with polyangiitis (Wegener): clinical aspects and treatment. Autoimmun Rev. 2014; 13: 1121-1125.
- Stone JH. Wegener's Granulomatosis Etanercept Trial Research Group. Limited versus severe Wegener's granulomatosis: baseline data on patients in the Wegener's granulomatosis etanercept trial. Arthritis Rheum. 2003; 48: 2299-2309.
- Rothschild PR, Pagnoux C, Seror R, Brezin AP, Delair E, Guillevinc L. Ophthalmologic manifestations of systemic necrotizing vasculitides at diagnosis: a retrospective study of 1286 patients and review of the literature. Semin Arthritis Rheum. 2013; 42: 507-514.
- Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992; 116: 488-498.
- Ahmed M, Niffenegger JH, Jakobiec FA, Ben-Arie-Weintrob Y, Gion N, Androudi S, et al. Diagnosis of limited ophthalmic Wegener granulomatosis: distinctive pathologic features with ANCA test confirmation. Int Ophthalmol. 2008; 28: 35-46.
- Bhatia A, Yadava U, Goyal JL, Chaturvedi KU. Limited Wegener's granulomatosis of the orbit: a case study and review of literature. Eye (Lond). 2005; 19: 102-104.
- Isa H, Lightman S, Luthert PJ, Rose GE, Verity DH, Taylor SR. Histopathological features predictive of a clinical diagnosis of ophthalmic granulomatosis with polyangiitis (GPA). Int J Clin Exp Pathol. 2012; 5: 684-689.
- Lamprecht P, Gross WL. Current knowledge on cellular interactions in the WG-granuloma. Clin Exp Rheumatol. 2007; 25: 49-51.
- Kamali S, Kasapoglu E, Aktürk F, Gül A, Inanc M, Ocal L, et al. Eosinophilia and hyperimmunoglobulinemia E as the presenting manifestations of Wegener's granulomatosis. Clin Rheumatol. 2003; 22: 333-335.
- Krupsky M, Landau Z, Lifschitz-Mercer B, Resnitzky P. Wegener's granulomatosis with peripheral eosinophilia. Atypical variant of a classic disease. Chest. 1993; 104: 1290-1292.
- Potter MB, Fincher RK, Finger DR. Eosinophilia in Wegener's granulomatosis. Chest. 1999; 116: 1480-1483.
- Yousem SA, Lombard CM. The eosinophilic variant of Wegener's granulomatosis. Hum Pathol. 1988; 19: 682-688.
- Chan AS, Yu DL, Rao NA. Eosinophilic variant of Wegener granulomatosis in the orbit. Arch Ophthalmol. 2011; 129: 1238-1240.
- Ntatsaki E, Carruthers D, Chakravarty K, D'Cruz D, Harper L, Jayne D, et al. BSR and BHPR guideline for the management of adults with ANCA-associated vasculitis. Rheumatology (Oxford). 2014; 53: 2306-2309.