
Research Article
Austin J Clin Immunol. 2022; 8(1):1046.
Propofol Protects Cardiac Myocytes from H2O2-induced Cell Injury via Regulating ROS Accumulation and Mitochondrial Function from Oxidative Stress via Regulating ROS and Mitochondria
Hou Z, Guo L and Zhang Q*
Department of Anesthesiology, The First Affiliated Hospital of the University of South China, Hengyang, Hunan Province, P.R. China
*Corresponding author: Qianlu Zhang, Department of Anesthesiology, The First Affiliated Hospital of the University of South China, Hengyang, Hunan Province, 421205, P.R. China
Received: January 27, 2022; Accepted: February 23, 2022; Published: March 02, 2022
Abstract
Oxidative stress is one of the main mechanisms of myocardial ischaemia-reperfusion (I/R)-induced injury, which is one of the main cause of cardiomyocyte death and precipitates life-threatening heart failure. Propofol (2,6-diisopropylphenyl, PR) plays critical roles, including in I/R-induced oxidative stress; however, its protective effects on I/R-induced oxidative stress are still largely unknown. Considering that oxidative stress strongly affects mitochondrial function, we hypothesized that propofol may regulate I/R-induced H9C2 injury by modifying mitochondrial function. In a H2O2-induced cell model, propofol treatment reversed the H2O2-induced blockade of the cell cycle at the G1 phase and promotion of apoptosis. Two-hour pretreatment promoted proliferation and inhibited apoptosis, indicating that propofol pretreatment may, decrease H2O2-induced ROS accumulation. Propofol decreased oxidative stress-induced haem oxygenase-1 (HO-1) expression, and ROS scavenging mediated by treatment with NAC also decreased HO-1 expression. Propofol decreased ROS accumulation after H2O2 treatment, which was similar to the effects of the ROS scavenger NAC. Further results also showed that propofol enhanced the maintenance of mitochondrial function. However, without affecting the mitochondrial DNA content, propofol decreased mitochondrial ATP production and transcriptional activity, indicating that propofol may temporarily block mitochondrial function to prevent oxidative stress. These results suggest that propofol protects cardiomyocytes by regulating mitochondrial function.
Keywords: Propofol; Oxidative stress; ROS; Cardiamyocyte; H9C2
Introduction
Ischaemia/reperfusion (I/R) injury is one of the most common types of damage during or after major surgeries, with the incidence of myocardial damage ranging from 1% to 7% [1,2]; myocardial damage is the main cause of death mediated by inducing oxidative stress and subsequent fatal damage to cardiomyocytes [2]. Reducing I/R injury is now one of the main strategies for achieving improved recovery [3], including ischaemic conditioning and preconditioning [4], herbal component conditioning [5], and chemical conditioning [6]. There are many factors that potentially contribute to protection against I/R-induced myocardial injury. However, the exact mechanism underlying myocardial injury induced by I/R is not completely understood; thus, no promising therapeutic candidates have been found. In this context, the protective effects of ischaemic conditioning and preconditioning on cardiomyocytes are gaining attention.
Propofol is a widely accepted and clinically used intravenous anaesthetic that also plays protective roles in oxidative stress induced cell injury by several factors in several kinds of cells, including cardiomyocytes [7], hippocampal neurons [8], and kidney cells [9]. Shinjo and colleagues presented that oxidative stress induced by H2O2 was decreased by propofol in rat cardiac H9C2 cells, which induced the transcriptional activity of antioxidant enzymes [7]. It is also reported inhibition of calcineurin-induced calcium overload and the subsequent activation of YAP signalling decreased hypoxiainduced oxidative stress in hippocampal neurons [7]. Propofol administration can protect the kidneys from sepsis-induced AKI via exerting inhibitory role on oxidative stress [10]. By considering that oxidative stress is one of the main cause in mitochondrial damage [11] and propofol treatment was reported to exert protective effects on hearts from reperfusion injury [12-14] it is worth investigating the role of propofol conditioning or preconditioning and potential molecular mechanisms on it protective effects.
As the major energy-producing organelle of the cell, the mitochondria produce a major proportion of reactive oxygen species (ROS) by metabolizing active oxidative species under oxidative stress conditions [15]. Excessive ROS can cause damage to mitochondrial energy production by damaging mitochondrial DNA (mtDNA) and disrupting the mitochondrial membrane potential [16]. Propofol has been reported to be tightly associated with mitochondrial biogenesis and function. Sumi reported that propofol disturbed mitochondrial function by blocking the mitochondrial electron transport chain, inducing a metabolic switch and causing cell death [17]. In hippocampal astrocytes and neurons, propofol promotes mitochondrial fission and mitophagy, which induce mitochondriadependent apoptosis [18]. In contrast, under oxidative stress induced by oxygen-glucose deprivation and reperfusion, propofol protects the mitochondrial ultrastructure and inhibits mitochondrial fission, but the exact molecular mechanism is unknown [19]. According to the literature, propofol tightly regulates mitochondrial function and homeostasis and results in context-dependent pro-apoptotic or prosurvival effects. These findings prompted us to investigate the exact roles of propofol in mitochondrial homeostasis and function under oxidative stress.
In present study, we focused on the regulatory roles of the intravenous anaesthetic propofol on mitochondrial homeostasis and function under oxidative stress induced by H2O2. Propofol exerts cytotoxic effects by disrupting mitochondrial function under normal conditions and exerts protective effects on the mitochondria under oxidative stress conditions [20]. We aimed to determine the exact effects of propofol on mitochondrial function and provide clinical suggestions for the use of propofol.
Material and Methods
Cell culture and treatment
Rat cardiomyocyte cell line H9C2 was bought from American Type Culture Collection (ATCC Manassas, VA, USA), and maintained in medium (DMEM) with addition of 10% Fetal bovine serum (FBS, Life Technologies, Grand Island, NY, USA), 100g/mL streptomycin and 100 U/mL penicillin. Cells were cultured in a humidified atmosphere of 5% CO2 and 95% air at 37oC and passaged every three days.
For inducing oxidative stress, 100, 200, 300, 400, 500, 600, 700, and 800 μM of H2O2, was supplemented into culture medium for 24h, respectively. For inducing proliferation-inhibiting effect, 243.5μM of H2O2 was employed for 24-hour incubation. For inducing apoptosispromoting effect, 425.1μM of H2O2 was employed for 24-hour incubation.
For assessing propofol’s effect, 50μM of propofol was employed for 24-hour incubation.
Cell cycle analysis
Propidium iodide (PI, Beyotime) followed by flow cytometry assay was employed for analyzing cell cycle distribution. Approximately 1×106 cells were fixed with ice-cold 75% ethanol for 12-16 h. Then, fixed cells were washed with ice-cold PBS twice and incubated with 100μg/ml RNase A and 40μg/ml PI in final concentration for 30min avoid from light. Then stained samples were investigated by Navios flow cytometers (Beckman Coulter, Brea, CA, USA). The proportion of G1/G0, S and G2/M phases were calculated by employing FlowJo 9.7.6 (FlowJo LLC, Ashland, OR USA).
Apoptotsis analysis
Cells were stained with Annexin V-FITC (green fluorescence) and the non-vital dye PI (red fluorescence). FITC+/PI- and FITC+/ PI+ proportions were considered as apoptotic cells, FITC-/PI+ proportion was considered as non-apoptotic cells. All samples were analyzed flow cytometry and data was analyzed using FlowJo 9.7.6 (FlowJo LLC, Ashland, OR USA).
ROS detection
Cells were treated with H2O2 for various duration, and 30 min prior to the termination of the treatment, 100ng/ml 2’,7’-dichlorofluorescein diacetate (H2DCFDA, Sigma-Aldrich, St. Louis, MO, USA) was added to the medium for 30-minute staining. Then cells were washed with ice-cold PBS twice and imaged under a X71 (U-RFL-T) fluorescence microscope (Olympus, Melville, NY).
Imaging of mitochondrial ROS
To identify mitochondrial ROS accumulation, cells were incubated with 2.5μM MitoSox Red for 15min and 10ng/ml DAPI was added simultaneously for nucleus staining. The excitation wavelength of MitoSox Red is 543nm and emission wavelength of 560-620 nm was collected. For each group, five views were randomly imaged under a X71 (U-RFL-T) fluorescence microscope (Olympus, Melville, NY).
Western blot
Samples were lysed by employing SoniConvert® Sonicator (DocSense, Chengdu, China) following the manufacture’s instruction. Protein concentration was measured using BCA kit (Sigma) and 20μg of total protein were separated by SDS-PAGE and transferred to PVDF membrane (Millipore) pre-blocked in 5% powdered milk+5% BSA in PBS containing 0.3% 20 for 30 min at room temperature (RT). Then, the blot-transferred membrane was incubated with primary antibody at 4oC overnight. Anti-Heme Oxygenase 1 antibody (Cat. No.: ab189491, Abcam) was diluted in 1:1000 and anti-beta Actin antibody (Cat. No: Ab8226, Abcam) was diluted in 1:5000. After washing for three times with PBS containing 0.3% Tween 20, the membrane was incubated with horseradish peroxidase (HRP)-labeled secondary antibody for 1h at RT. Then membrane was then developed by using ECL detection systems (Thermo Scientific, Waltham, MA, USA).
JC-1 staining
Cells were stained using 20μg/mL of JC-1 at 37°C for 20min. Then supernatant was removed and cells were washed using chilled PBS for two times. Cells were imaged. The red signal represents JC-1 aggregates, and green signal represents JC-1 monomers.
RT-qPCR
It was performed to detect mitochondrial-coded genes. Briefly, total RNA was extracted from 1×106 cells for each sample by using SoniConvertTM system (DocSense, Chengdu, China). Reverse transcription was performed by employing 1μg of total RNA.
For each reaction, 0.2μl of cDNA was employed. The reaction procedure was described as follows: 95h 5min, 35 cycles of 95h 10s, and 60h 1min. GAPDH forward: 5’-C C T T C A T T G A C C T C A A C T A C A T-3’; reverse: 5’-C C A A A G T T G T C A T G G A T G A C C-3’. GAPDH was considered as a internal control. COX I forward: 5’- G G A G C A G T A T T C G C C A T C A T-3’; reverse: 5’-G A G C A C T T C T C G T T T T G A T G C-3’; HO-1 forward: 5’-A A G A C T G C G T T C C T G C T C A A C-3’; reverse: 5’-A A A G C C C T A C A G C A A C T G T C G-3’; ND1 forward: 5’- C A C C C C C T T A T C A A C C T C A A-3’; reverse: 5’-A T T T G T T T C T G C G A G G G T T G-3’.
Statistical analysis
All data were analyzed for statistical significance using SPSS 13.0 software (SPSS, Chicago, IL, USA) and presented as mean±SD from at least 3 independent experiments performed in duplicate. Statistical comparisons of the results were made using analysis of One-way ANOVA followed by Tukey’s post-hoc test. P<0.05 was considered statistically significant.
Results
Propofol treatment exerts protective effect against H2O2-induced inhibition of cell proliferation. To determine the exact concentration of H2O2 in inducing inhibition of cell proliferation and cell death, a range concentration of H2O2 (100-800 μM) was co-incubated with H9C2 cells for 24 hours, and then assessed for cell viability. As it is shown in Figure 1A, continuous decrease of cell viability was observed, and 30% inhibitory concentration (IC30), approximately 243.5μM was employed in the term of proliferating inhibition, 50% inhibitory concentration (IC50), approximately 425.1μM was employed in the term of apoptosis promotion. To confirm that IC30 of propofol inhibits proliferation, but not stop proliferation, its effect on cell viability from day 1 to 4 was measured and it is observed that H9C2 cells cultured with 2O2 presented a slower proliferating rate compared to mock group (solvent control group) (Figure 1B). As a cell marker response to oxidative stress, heme oxygenase-1 (HO-1), a critical antioxidant enzyme that can be induced by oxidative stress [21], was detected by western blot. Expectedly, 2O2 (≥200μM in concentration) significantly upregulated HO-1 protein levels (Figure 1C). Aim to confirm whether propofol exerts protective effect on H2O2-induced cell injury, 10-100 μM of propofol was added in with IC30 of H2O2, and cell cycle phases were measured 24-hour later. As it is shown in figure 1D, expectedly, H2O2 treatment remarkably increased the proportion of G1/G0 phase, which is potentially the main cause of cell cycle arrest by H2O2. Notably, addition of both 50 and 100 μM of propofol significantly reversed the increase of G1/ G0 proportion induced by H2O2, indicated that propofol treatment decreased H2O2-induced inhibitory effect on proliferation. By considering that propofol alone presented no detectable effect on cell proliferation after 24h (data not shown), reverse of H2O2-induced cell cycle arrest by propofol may due to the block of oxidative stress.
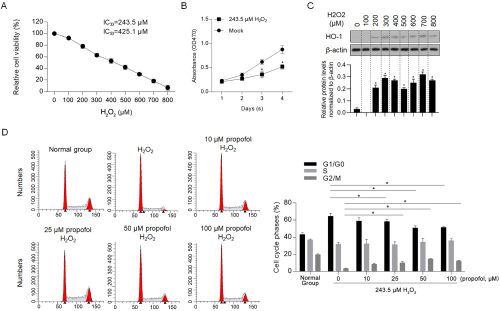
Figure 1: Propofol reversed H2O2-induced cell cycle arrest.
A) Cell cytotoxicity of H2O2 was measured by performing CCK-8 assay. B) The effect of IC30 of H2O2 on proliferation from day 1 to 4 in H9C2 cells was measured by performing CCK-8 assay. *P<0.05, vs. mock group. C) Western blot was performed to detect the protein level of HO-1 after H2O2 treatment for 24h. D) Different concentration of propofol and IC30 of H2O2 were supplemented in medium for 24-hour culturing, then cells were stained with PI and analyzed by flow cytometry. *P<0.05, vs. 243.5μM H2O2/0μM propofol group.
Propofol decreased H2O2-induced apoptosis
It is presented that, 10-100 μM of propofol presented no detectable effect on apoptosis (data no shown), this indicated that propofol treatment may target to block H2O2-induced oxidative stress. By considering this, we hypothesized that pretreatment of propofol may achieve better effects on exerting protective effect. H9C2 cells pretreated with 50μM of propofol for 0.5, 1, 2, and 4 hours were then exposed to IC50 of H2O2. Cell viability was observed to be higher in 2- and 4-hour pretreated groups (Figure 2A). To evaluate the effect of propofol on H2O2-induced apoptosis, IC30 of H2O2 and different concentration of propofol were added for co-incubation for 24 hours, then the apoptosis was measured. As it is shown in Figure 2B and 2C, H2O2 significantly induced apoptosis after 24-hour incubation. Addition of both 50 and 100 μM of propofol significantly decreased apoptotic proportion induced by H2O2, indicated that propofol may reverse oxidative stress induced by H2O2. In further experiments, 50μM of propofol was used. To further confirm this observation, all cells were then analyzed by apoptotic detection. Expectedly, propofol treatment decreased apoptosis after H2O2 exposure (Figure 2D and 2E), and 2- and 4-hour pretreated groups presented much less proportion of apoptotic cells.
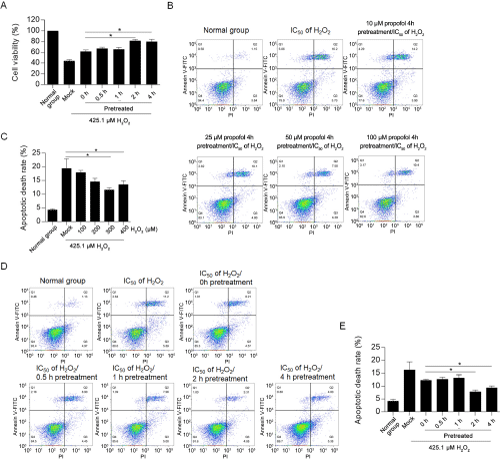
Figure 2: Pre-treatment of propofol exerts protective effects on H2O2-induced apoptosis.
To evaluate the protective effect of pre-treatment of propofol, 0.5-4h pretreated H9C2 cells were then incubated with IC50 of H2O2 for 24h, followed by performing
CCK-8 assay (A) and flow cytometry (B&C). *P<0.05, vs. H2O2 treated group. D&E. 425.1μM of H2O2 and 50μM of propofol were co-incubated with H9C2 for 24 hours, and then cells double stained with Annexin V-FITC and PI were analyzed by flow cytometry. *P<0.05, vs. H2O2 treated group.
To find out whether 50μM of propofol exerts protective effect, neonate cardiomyocytes were isolated and cultured. 2-day-old newborn rat was sacrificed for isolating rat cardiac myocytes (RCM). Isolated cells were cultured in 6-well plate at different confluence (Figure 3A). After 24h treatment under IC30 of H2O2, cell viability and cell cycle phases were measured. As presented in figure 3B, expectedly, H2O2 treatment decreased cell viability, which was reversed by treatment of propofol. Cell cycle distribution was also beening arrested at G1/G0 phase after H2O2 addition, which was reversed by propofol (Figure 3C). The protective effect of propofol on oxidative stress-induced apoptosis in RCM was then measured by Annexin V-FITC/PI double staining assay. As shown in Figure 3D, IC50 of H2O2 significantly induced apoptosis, which was reversed by addition of propofol?
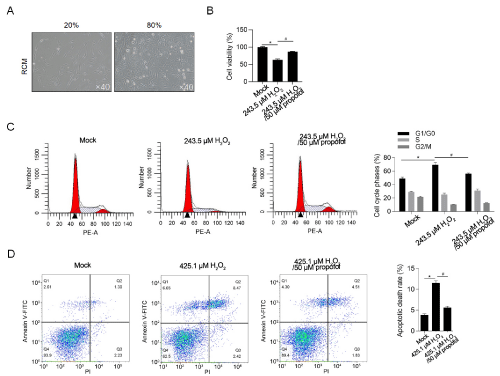
Figure 3: Pre-treatment of propofol exerts protective effects on oxidative stress in RCM.
A) Morphology of isolated RCM at different confluence. B) After being cultured in 243.5μM of H2O2, with or without 50μM of propofol, cell viability was measured by cck-8 assay. *P<0.05, vs. Mock group. #P<0.05, vs. H2O2 group. C) After being cultured in 243.5μM of H2O2, with or without 50μM of propofol, cell cycle phases were measured by performing PI staining followed by flow cytometry. *P<0.05, vs. Mock group. #P<0.05, vs. H2O2 group. D) After being cultured in 425.1μM of H2O2, with or without 50μM of propofol, cell apoptosis was measured by performing Annexin V-FITC/PI double staining followed by flow cytometry. *P<0.05, vs. Mock group. #P<0.05, vs. H2O2 group.
Propofol exerts protective effect on H2O2-induced cell injury potentially via inhibiting ROS accumulation
By considering that H2O2-induced toxicity is mainly via promoting ROS accumulation [22], we wondered that whether pretreatment of propofol on H2O2-induced oxidative stress also modify ROS accumulation. ROS staining results presented that, after H2O2 exposure, ROS accumulation was observed (Figure 4A). In all propofol treated groups, ROS accumulation was obviously decreased, compared to untreated group, and pretreatment further decreased ROS level, indicated that propofol pretreatment may inhibit ROS accumulation induced by H2O2-induced oxidative stress. To further confirm whether the modification of ROS accumulation by propofol is critical for its protective effect under oxidative stress induced by H2O2, the effects of propofol with or without NAC, a ROS scavenger, on proliferation and apoptosis were accessed. As it is shown in Figure 4B and 4C, both NAC and propofol treatment reversed H2O2-induced proliferating inhibition and apoptosis promotion, and co-treatment of NAC and propofol failed to exert further modification on these two processes, indicated that propofol may exert protective effect mainly via decrease ROS level.
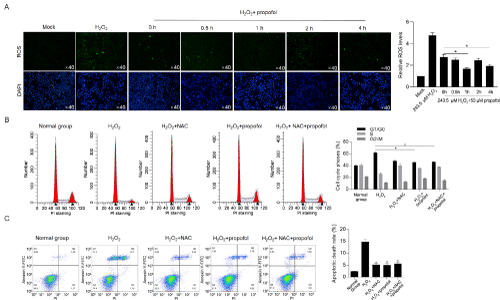
Figure 4: Propofol abolishes H2O2-induced ROS accumulation and thus exerts protective effects. A) Intracellular ROS levels were determined by H2DCFDA staining and imaged. B) After 10μM of NAC, 50μM of propofol, or NAC/propofol treatment with 243.5μM of H2O2 exposure, cell cycle phases were detected by PI staining followed by flow cytometry. *P<0.05, vs. H2O2 group. C) After 10μM of NAC, 50μM of propofol, or NAC/propofol treatment with 425.1μM of H2O2 exposure, apoptosis rates were detected by Annexin V-FITC/PI double staining followed by flow cytometry. *P<0.05, vs. H2O2 group.
Propofol may protect H9c2 cells from oxidative stress via presence of HO-1
By considering that HO-1 is crucial for antioxidantation after oxidative stress, we hyperthized that propofol may protect cardiac myocytes via HO-1 directly or indirectly. By performing RT-qPCR and western blot, it was observed that H2O2 treatment expectedly increased HO-1 expression (Figure 5A and 5B). Both addition of NAC (a ROS scavenger) and propofol (at final concentration of 50μM) significantly decreased HO-1 expression, indicated that propofol may regulates HO-1 expression via decreasing ROS accumulation. We then efficiently knockdown HO-1 expression by transfecting shRNA target to HO-1 mRNA (Figure 5C), and detected ROS accumulation. Expectedly, propofol treatment significantly decreased ROS level only when HO-1 was presented (Figure 5D). By detecting cell cycle arrest and apoptosis induced by oxidative stress, it was clearly observed that propofol only exerted protective effects on cell cycle and cell survival when HO-1 was presented (Figure 5E and 5F).
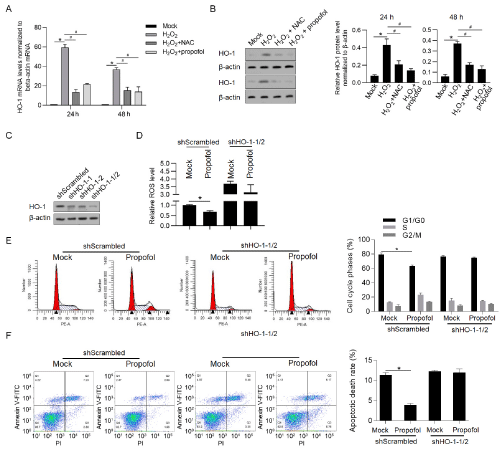
Figure 5: Propofol potentially exerts protective effects via regulating HO-1 expression.
After treatment of 10μM of NAC, 50μM of propofol, HO-1 mRNA (A) and protein (B) levels were detected. *P<0.05, mock group; #P<0.05, H2O2 group. C) After
tranfection of shRNA target to HO-1 mRNA, western blot was measured to detect HO-1 protein level. After HO-1 knockdown, the effects of propofol on ROS
accumulation (D), cell cycle arrest (E) and cell apoptosis (F) were measured. *P<0.05, mock group.
Propofol treatment potentially decreased mitochondrial function and maintained mitochondria function
We also detect mitochondrial ROS by performing mitotracker SoxRed after propofol treatment. Expectedly, H2O2-induced increase in mitochondrial ROS was obviously decreased by 0.5- and 2-hour pretreatment of propofol (Figure 6A). H2O2-induced oxidative stress causes cellular injury mainly via inducing loss of mitochondrial homeostasis [22], this promoted us to confirm whether propofol affects mitochondrial membrane potential by performing JC-1 staining. As it is presented in Figure 6B, propofol treatment obviously reversed H2O2-induced loss of mitochondrial membrane potential. Without affecting mitochondrial DNA contents (Figure 6C), propofol treatment decreased mitochondrial ATP synthesis (Figure 6D) and mitochondrial transcriptional activity (Figure 6E). Taken together, propofol treatment decreased mitochondrial function, which may due to the decrease in ROS accumulation.
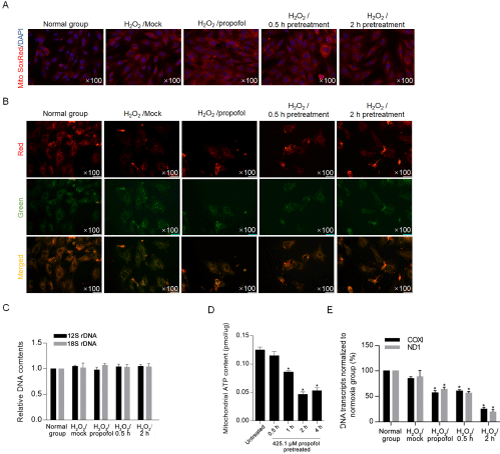
Figure 6: Propofol decreased mitochondrial ROS and maintained mitochondrial homeostasis.
A) After propofol pretreatment, mitochondrial ROS induced by H2O2 was detected by performing mitoSoxRed staining. B) To determine mitochondrial homestasis,
JC-1 staining was performed. C) To determine mitochondrial DNA contents, the relative DNA contents of 12S rDNA and 18S rDNA were measured by quantitative
PCR. Mitochondrial ATP synthesis (D) and mitochondrial transcriptional activity (E) were analyzed. *P<0.05, vs. H2O2 group.
Discussion
In this report, we demonstrated the regulatory effects of propofol on mitochondrial mass, ATP production, transcriptional activity and membrane potential to limit oxidative stress induced by H2O2. Propofol treatment and preconditioning maintained mitochondrial function while exerting suppressive effects on mitochondrial ATP production and mitochondrial transcriptional activity. In rat H9C2 cells, propofol reversed the inhibition of proliferation and promotion of apoptosis mediated by H2O2-induced oxidative stress, indicating a potential protective mechanism involving protection of the mitochondria.
Propofol has been shown to have antioxidant and antiapoptotic effects [23]. In human umbilical vein endothelial cells (HUVECs), propofol exerts antioxidant and pro-survival effects against H2O2-induced oxidative stress. Propofol treatment attenuated the activation of caspase-3 and expression of Bcl-2 and Bax in a dosedependent manner. Propofol also ameliorated the H2O2-induced phosphorylation of p38 MAPK, JNK, and Akt signaling pathway components, which are critical for initiating or promoting apoptosis [24]. Furthermore, propofol exerts protective effects against H2O2 induction. Takabuchi used a hypoxia model to illustrate the effects of propofol on nontumour liver Hep3B cells and showed that hypoxia-induced factor 1 (HIF-1) was transcriptionally and actively inhibited by propofol; thus, propofol exerted a protective effect against hypoxia exposure. The exact mechanism by which propofol regulates HIF-1 and its downstream signaling pathway, including lactate dehydrogenase and vascular endothelial growth factor pathways, and promotes cell survival under hypoxic stress is not known [25]. Zhao and colleagues used oxygen-glucose deprivation and reperfusion models established with H9C2 cells and showed that propofol treatment exerted cardioprotective effects by increasing FoxO1 expression in a dose-dependent manner and inhibiting p-AMPK generation [26]. However, how propofol exerts protective effects against oxidative stress-induced injury in the mitochondria is still worth to be investigated.
We revealed that both treatment with propofol and pretreatment with propofol obviously decreased the ROS accumulation induced by H2O2, indicating that propofol exerts antioxidant effects. The accumulation of ROS, which are mainly produced in the mitochondria, is the main cause of oxidative stress and leads to mitochondrial DNA damage and loss of the mitochondrial membrane potential [16]. Oxidative stress induced by H2O2 leads to ROS accumulation, which has been reported to be negatively associated with the mitochondrial membrane potential [27], and these findings prompted us to examine whether propofol treatment regulates mitochondrial function. Although H2O2-induced oxidative damage marker, such as p-histone (Ser139-H2A) was not detected which is a potential limitation of the present study, the H2O2-induced cell damage was detected by performing CCK-8 assay and Annexin V-FITC/PI double staining followed by flow cytometry. As expected, propofol treatment helped to maintain function in the context of oxidative stress induced by H2O2. This result demonstrated that propofol treatment helped to maintain mitochondrial function by decreasing the ROS accumulation induced by H2O2. Although the molecular mechanism remains largely unknown, we hypothesized that propofol may maintain mitochondrial homeostasis by promoting mitochondrial biogenesis. In further study, it is worth investigating the exact molecular mechanism of propofol function on pretecting mitochondrial homeostasis by several experimental approaches, such as detecting JC-1 staining and mitochondrial-induced ROS.
We further found that propofol conditioning markedly decreased mitochondrial function by decreasing ATP production and decreased mitochondrial transcriptional activity without affecting mitochondrial DNA levels. As a limitation, we failed to perform Seahorse XFp Analyzer, which is powerful tool for the assessment of cellular respiration, which is a limitation in this study. The mitochondria play key roles in inducing apoptotic cell death via ROS-induced pathways [28], including the regulation of calcium levels and the opening of the mitochondrial permeability transition pore, followed by the release of cytochrome. It is reported that propofol interacts with mitochondrial complexes I, II, and III and subsequently causes a cellular metabolic switch from oxidative phosphorylation to glycolysis [28]. Furthermore, generated ROS activates caspases 9 and 3/7 and induce mitochondria-dependent apoptosis. According to our data, propofol treatment decreased mitochondrial function, which may be the cause of the decrease in ROS levels, and maintained mitochondrial function under oxidative stress. This finding demonstrates that propofol mediates protective effects rather than promoting apoptosis.
Conclusion
In summary, propofol suppresses mitochondrial function, maintains mitochondrial homeostasis, inhibits ROS accumulation, and exerts a protective effect under oxidative stress induced by H2O2. Our data also demonstrated that propofol pretreatment exerted regulatory effects on the mitochondria and cellular injury induced by oxidative stress. These results provide new insight into the regulatory roles of propofol in the mitochondria under oxidative stress.
Declaration
Acknowledgements: The author would like to thank Dr. Dongsheng Liao (Third Military Medical University, Chongqing) for language editing and help.
Funding: This work was financially supported by the National Natural Sciences Foundation of China (No. 81870337).
Availability of data and materials: The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- Landesberg G, Beattie WS, Mosseri M, Jaffe AS, Alpert JS. Perioperative myocardial infarction. Circulation. 2009; 119: 2936-2944.
- Mauermann E, Puelacher C, Lurati BG. Myocardial injury after noncardiac surgery: an underappreciated problem and current challenges. Curr Opin Anaesthesiol. 2016; 29: 403-412.
- Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013; 123: 92-100.
- Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003; 83: 1113-1151.
- Yu H, Shi L, Qi G, Zhao S, Gao Y, et al. Gypenoside Protects Cardiomyocytes against Ischemia-Reperfusion Injury via the Inhibition of Mitogen-Activated Protein Kinase Mediated Nuclear Factor Kappa B Pathway In Vitro and In Vivo. Front Pharmacol. 2016; 7: 148.
- Chen Y, Wang L, Zhang L, Chen B, Yang L, et al. Inhibition of Connexin 43 Hemichannels Alleviates Cerebral Ischemia/Reperfusion Injury via the TLR4 Signaling Pathway. Front Cell Neurosci. 2018; 12: 372.
- Shinjo T, Tanaka T, Okuda H, Kawaguchi A, Oh-Hashi K, et al. Propofol induces nuclear localization of Nrf2 under conditions of oxidative stress in cardiac H9c2 cells. PLoS One, 2018; 13: e196191.
- Li X, Yao L, Liang Q, Qu H, Cai H. Propofol Protects Hippocampal Neurons from Hypoxia-Reoxygenation Injury by Decreasing Calcineurin-Induced Calcium Overload and Activating YAP Signaling. Oxid Med Cell Longev. 2018; 2018: 172-191.
- Zheng G, Qu H, Li F, Ma W, Yang H. Propofol attenuates sepsis-induced acute kidney injury by regulating miR-290-5p/CCL-2 signaling pathway. Braz J Med Biol Res. 2018; 51: e7655.
- Hosing CH, Chou W, Wang JJ, Chen HW, Yeh CH. Propofol increases bone morphogenetic protein-7 and decreases oxidative stress in sepsis-induced acute kidney injury. Nephrol Dial Transplant. 2011; 26: 1162-1172.
- Huang ML, Chiang S, Kalinowski DS, Bae DH, Sahni S, et al. The Role of the Antioxidant Response in Mitochondrial Dysfunction in Degenerative Diseases: Cross-Talk between Antioxidant Defense, Autophagy, and Apoptosis. Oxid Med Cell Longev. 2019; 2019: 6392763.
- Javadov SA, Lim KH, Kerr PM, Suileiman M, Angelini G, et al. Protection of hearts from reperfusion injury by propofol is associated with inhibition of the mitochondrial permeability transition. Cardiovasc Res. 2000; 45: 360-369.
- Deng F, Wang S, Zhang LQ, Zhang L, Xie X, Cai S, et al. Propofol through upregulating caveolin-3 attenuates post-hypoxic mitochondrial damage and cell death in H9C2 cardiomyocytes during hyperglycemia. Cell Physiol Biochem. 2017; 44: 279-292.
- Zahgn QL, Cai SW, Guo LP, Zhao G. Propofol induces mitochondrialassociated protein LRPPRC and protects mitochondria against hypoxia in cardiac cells. PLoS One. 2020; 15: e0238857.
- Napier I, Ponka P and Richardson DR. Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood. 2005; 105: 1867-1874.
- Cha MY, Kim DK, Mook-Jung I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp Mol Med. 2015; 47: e150.
- Sumi C, Okamoto A, Tanaka H, Nishi K, Kusunoki M, et al. Propofol induces a metabolic switch to glycolysis and cell death in a mitochondrial electron transport chain-dependent manner. PLoS One. 2018; 13: e192796.
- Liu Y, Yan Y, Inagaki Y, Logan S, Bosnjak ZJ, et al. Insufficient Astrocyte- Derived Brain-Derived Neurotrophic Factor Contributes to Propofol-Induced Neuron Death Through Akt/Glycogen Synthase Kinase 3beta/Mitochondrial Fission Pathway. Anesth Analg. 2017; 125: 241-254.
- Wang H, Zheng S, Liu M, Jia C, Wang S, et al. The Effect of Propofol on Mitochondrial Fission during Oxygen-Glucose Deprivation and Reperfusion Injury in Rat Hippocampal Neurons. PLoS One. 2016; 11: e165052.
- Kido K, Ito H, Yamamoto Y, et al. Cytotoxicity of propofol in human induced pluripotent stem cell-derived cardiomyocytes. J Anesth. 2018; 32: 120-131.
- Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J, et al. Role of Nrf2/ HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. 2016; 73: 3221-3247.
- Ni T, Yang W, Xing Y. Protective effects of delphinidin against H2O2-induced oxidative injuries in human retinal pigment epithelial cells. Biosci Rep. 2019; 39.
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014; 94: 909-950.
- Marik PE. Propofol: an immunomodulating agent. Pharmacotherapy. 2005; 25: 28S-33S.
- Xie CL, Pan YB, Hu LQ, Qian YN. Propofol attenuates hydrogenperoxideinduced apoptosis in human umbilical vein endothelial cells via multiple signaling pathways. Korean J Anesthesiol. 2015; 68: 488-495.
- Takabuchi S, Hirota K, Nishi K, Oda S, Oda T, et al. The intravenous anesthetic propofol inhibits hypoxia-inducible factor 1 activity in an oxygen tension-dependent manner. FEBS Lett. 2004; 577: 434-438.
- Zhao D, Li Q, Huang Q, Li X, Yin M, et al. Cardioprotective effect of propofol against oxygen glucose deprivation and reperfusion injury in H9c2 cells. Oxid Med Cell Longev. 2015; 2015: 184938.
- Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997; 416: 15-18.