
Research Article
Austin J Clin Med. 2014;1(1): 1005.
Identifying a Specific Biomarker Allows for Improved Quality of Life
Vincent Cracolici1, Victor Elgabalawi1 and Houria I Hassouna1,*
1College of Human Medicine, Michigan State University, USA
*Corresponding author: Houria I Hassouna, College of Human Medicine, Michigan State University, B-214 Clinical Center, 788 Service Road East Lansing, MI 48824-1313, USA
Received: January 02, 2014; Accepted: February 10, 2014; Published: February 14, 2014
Abstract
Serositis and intermittent fever with cyclic white cell count elevations above normal range pose diagnostic challenges. Identifying specific biomarkers may allow for earlier treatment and improved quality of life for those diseases that share common symptoms and fall ambiguously in the domain of the rheumatologist, the infectious disease specialist or the hematologist. As our approach to diagnostics is growing increasingly sophisticated and moleculartools are becoming more commonplace, genotype-phenotype correlations should become immediately clinically useful.
Keywords: Diagnostic challenge; Specific biomarker; Cyclic leukocytosis; Intermittent fever; Familial Mediterranean Fever; Autoinflammatory syndromes; Pyrin; MEFV gene mutations.
Introduction
Serositis and intermittent fever with cyclic white cell count elevations above normal range pose a diagnostic challenge to the rheumatologist, the infectious disease specialist or the hematologist (Table 1).
![]()
Medical Specialty
Syndromes and diseases characterized by intermittent fever and cyclic leukocytosis
Rheumatology
- Juvenile rheumatoid arthritis
- Systemic lupus erythematosus
- Periodic fever Syndromes
- Familial Mediterranean Fever
Infectious Disease
- Lyme disease
- Tuberculosis
- Congenital Syphilis
Hematology
- Acute and chronic myeloid leukemia
- Acute and chronic lymphocytic leukemia
- Hodgkin’s lymphoma
Table 1: Autoimmune and autoinflammatory syndromes infectious and hematologic groups of diseases characterized by cyclic leukocytosis and intermittent fever.
These common symptoms may pique suspicion, particularly in children and adolescents, for the autoimmune diseases juvenile rheumatoid arthritis [1-3] and lupus erythematosus [4,5], they are encountered in Lyme disease from infection with Borrelia burgdorferi infection [6,7], in reactions adverse to [8], and in the delayed responses to the Treponema pallidum bacteria in congenital syphilis [9-11] Episodes of fever and involvement of skin, serous membranes, eyes, joints, gastrointestinal tract, and nervous system, predominantly with a childhood onset are characteristic of twelve known monogenic autoinflammatory syndromes [12]. In the autoinflammatory syndromes, aberrant inflammasome activation riginates from innate immune system dysregulation [13] Identifying the underlying cause for the common symptoms based on clinical findings alone is difficult. Pathologic changes underlying the common symptoms are characterized by unique biomarkers. Biomarkers are anatomic, physiologic, biochemical, molecular, or genetic parameters associated with the presence and severity of specific diseases and are detectable in diagnostic panels by a variety of methods, including physical examination, laboratory assays, imaging and gene probes [14]. Spectral karyotype identify numerical and structural markers of chromosome abnormalities. Biomarkers of autoimmunity are conspicuously absent in the infectious and inherited febrile joint and muscle diseases [15].
From our referral file we selected a case study describing an individual with symptoms typically associated with rheumatologic, infectious, and hematologic disorders and we dwell on the value of selecting the distinctive biomarker capable of differentiating one disease entity from others.
Case study
The individual, a middle aged white female, was referred for investigation of chronically elevated white cell counts above the normal range. Permission from the patient was granted to describe the case. Born in 1959 she is of Ashkenazi Jewish heritage and she was frequently sick at 6 years old with low grade fevers, intermittent joint pain, and stiffness in knees with that made her too ill to attend school. A persistence of childhood symptoms for 15 years past adolescence was associated with episodic anemia, lymphocytosis, and mild thrombocytopenia that the pediatrician attributed to Epstein Barr virus but theinfectivity was never confirmed. She was diagnosed with manic depressive bipolar disorder at age 20 and began lithium treatment. Her obesity was controlled by bariatric surgery at age 44. Her mother is in excellent health, father died from lung cancer, younger sister was diagnosed at 14 years of age with rare brain tumor treated with radiation, and older sister had a mature ovarian cystic teratoma removed at age 19.
Laboratory data
To aid the search for a diagnosis, leukemia and preleukemic syndrome were investigated when she was 20 years old. Histologic and flow cytometry examination of bone marrow biopsies did not show evidence of megakaryocytosis, abnormal infiltrates, or abnormal cell maturation. The Philadelphia chromosome, a translocation between chromosomes 22 and 9, was not detected in the spectral karyotype analysis. Flow cytometry results on peripheral blood were unremarkable. Autoimmune disorder was ruled out by reports of negative test results for antinuclear antibody, anti-DNA antibody, anti- smooth muscle antibody, and anti- mitochondrial antibody. The unexplained leukocytosis was rediscovered in 1995 blood counts to monitor her lithium therapy when she was 36 years of age. In Table 2, bimonthly leucocyte counts taken from CBC test results in years 1995, 1997, 1999, 2000, 2005 are presented. Although no data was found for year 1998, and data presented for 1996, 2001, 2002 and 2004 is incomplete, test results compiled in Table 2 demonstrate aclear pattern of cyclic leukocytosis with counts reaching above 13 billion per Liter in 5 instances, two of them occurring in 1995. At 8.4 billion per Liter the lower counts are significantly above the normal range of 1.6 to 7.2 billion per Liter.
![]()
Leukocyte Count (in billion/liter) from 1995-2005
1995
1996
1997
1999
2000
2001
2002
2003
2004
2005
11.0
10.9
9.2
9.1
13.4
11.4
10.5
8.8
10.5
13.2
13.4
11.4
11.4
9.1
8.4
8.7
11.2
10.8
12.1
10.9
12.4
11.0
10.9
11.4
8.9
11.4
11.4
13.1
11.0
10.0
9.0
9.1
11.2
11.4
13.4
12.5
17.1
13.4
12.9
9.8
9.1
10.5
8.4
10.5
12.2
10.9
12.3
10.9
8.8
10.1
Table 2: Leukocyte Count from CBC laboratory results (1995 to 2005). Values represent leukocyte counts (in billion/liter) from CBC required in the management of lithium therapy for an unrelated psychiatric condition from 1995-2005. The counts go as high as 13.4 billion/liter and never return to baseline, remaining elevated at 8.4 billion/liter. Normal ranges (1.6-7.2 billion/liter).
In Table 3 uric acid and micro-albumin to creatinine ratio test results confirm previous findings in year 2003 of mild renal impairment. C -reactive protein a biomarker of chronic inflammation is measured by highly sensitive antibody assay. Result at 0.2mg/dL is elevated above the upper limit of the normal range (0.08 mg/dL).
![]()
Laboratory test
Concentration
Normal range
Significance
Vitamin D
16ng
15.0-57.0ng
Within normal range
C-peptide
4.7ng
0.5-5.0 ng
Marker for high levels endogenous insulin after high glucose intake or insulin resistance
RBC folate
556 ng/mL
< 281 ng/mL
Adequate
Hematocrit
42.3%
35.4-44.2%
Within normal range
Pre-albumin
39.1 mg/dL
18-45 mg/dL
Within normal range
Cholesterol
187 mg/dL
<200 mg/dL
Within normal range
Triglyceride
252 mg/dL
<200 mg/dL
Elevated
Low Density Lipoprotein
78 mg/dL
<100 mg/dL
Within normal range
Uric Acid
6.9 mg/dL
2.4-6.0 mg/dL
Elevated
TSH
1.30 IU/mL
0.5-5.0 IU/mL
Within normal range
Hemoglobin A1C
5.5%
3.5-6.0
Borderline elevated
Vitamin B-12
490 pg/mL
271-870
Within normal range
Microalbumin: creatinine ratio
74 mg/dL
3-30 mg/dL
Elevated
C-reactive protein
0.2 mg/dL
0-0.08 mg/dL
Elevated
Table 3: Laboratory Data from 1 year prior to referral. The table illustrates the laboratory findings significant for elevated triglycerides, elevated uric acid, and elevated micro-albumin to creatinine ratio. C-reactive protein a biomarker of chronic inflammation was elevated at 0.2mg. Elevated C-reactive protein and elevated microalbumin were first identified when she had a gastric bypass in 2003. They were interpreted as indicating early renal damage as well as a chronic inflammatory condition.
In figure 1 reproduced with permission, C-reactive protein also measured by highly sensitive antibody assays in pediatric inflammatory bowel disease, patients with measurable standard C-reactive protein levels between 0.01 and 1 mg/L had active disease [16].
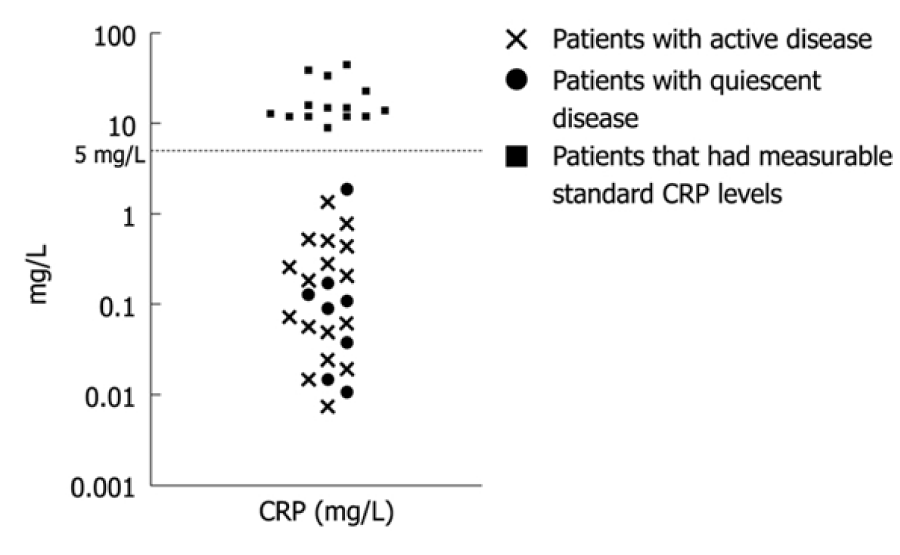
Figure 1: Reproduced with permission, C-reactive protein measured by highly sensitive antibody assays in peadiatric inflammatory bowel disease, patients with measurable standard C-reactive protein levels between 0.01 and 1 mg/L had active disease [16].
Tentative diagnosis
We considered familial Mediterranean Fever for the lifelong fluctuating leukocyte counts, chronic cyclic joint pain and stiffness, intermittent fever, anemia, elevated acute phase response C-reactive protein, absence of hematopoietic disorder, and elevated microalbumin to creatinine ratio indicative of altered kidney function. Familial Mediterranean Fever is an autoinflammatory disorder, one of four monogenic periodic fever disorders [17]. Nonetheless, at age 44 years, her European heredity, her lifelong symptoms, and the biochemical and molecular parameters detected in the laboratory, none characterize familial Mediterranean Fever to the exclusion of all other disorders.
Clinical suspicion of familial Mediterranean Fever remains closely coupled with a history of acute serosal episodes, ethnicity, and a positive family history [18]. Physical exam may demonstrate febrile symptoms during an attack, however patients are typically asymptomatic between episodes. Patients are often misdiagnosed with appendicitis due to presentation with an acute abdomen [19]. Laboratory studies are also nonspecific, with elevated levels of acute phase plasma proteins and possible proteinuria secondary to amyloidosis depending on the age of the patient and relative to time elapsed since the last episode. Erysipelas-like skin symptoms as well as Henoch-Schonlein purpura-like vasculidities have also been described in familial Mediterranean Fever [20-22].
Unique Biomarkers
In our case study, familial Mediterranean Fever diagnosis was confirmed by the finding of two polymorphisms, E148Q and V726A, previously characterized as disease-associated alleles [22-24]. Her mother carries both polymorphic E148Q and V726A alleles and the inherited E148Q allele from her father distinguishes her as carrier of the disease. Both E148Q and V726A polymorphisms have been associated with a more mild, waxing and waning form of familial Mediterranean Fever with amyloidosis occurring later in life relative to other mutations [23], however, it is highly likely that there are numerous environmental and genetic modifying factors associated with alterations in phenotype [25-27]. This case study illustrates the concept of genotype-phenotype correlation within familial Mediterranean Fever, and aligns with current epidemiological data that suggests numerous different clinical presentations of familial Mediterranean Fever due to various interactions of different alleles as well as interactions on an epigenetic level [26,27]. Specific genotypephenotype relationships have already been elucidated particularly regarding differential presence and severity of amyloidosis within the E148Q allele [26,28]. The genotype present in this patient likely explains the somewhat atypical presentation without the classic abdominal and serosal symptoms. Inheritance of E148Q from twoparents with good renal function may explain the relatively late onset of her mild renal impairment. She was prescribed colchicine 0.5 mg tablets twice daily that she claimed she could not tolerate and she requested a referral to another academic medical center. Repeattesting of the patient’s pyrin gene confirmed both polymorphisms and she was advisedto take colchicine and again she abstained. Her psychiatric condition constitutes a major concern. In regular reports from her nephrologist her kidney functions remain stable. The possible relationship between non-target pharmacologic alterations of FMF symptoms, perhaps with long term lithium therapy, as potentiation or attenuation, does not appear to be detailed in the literature and certainly merits further study.
Analysis and Interpretation
Epidemiology
Classic familial Mediterranean Fever is a monogenic autoinflammatory disorder occurring almost exclusively among ethnic groups of the Mediterranean basin [12,29]. A population based survey reveals an extremely high familial Mediterranean Fever carrier incidence of 1:3 in Armenia with a prevalence estimated to be 1:500 in Armenians. In Sephardic Jews the prevalence of FMF is estimated to be 1 per 250 to 1 per 1,000, with a much lower carrier incidence (1 per 73,000) in Ashkenazi Jews. Familial Mediterranean Fever is also found in Arabs, Greeks, Cubans, Belgians, and in Sweden, Germany, Korea, Japan, with a worldwide prevalence estimated at 100,000 - 150,000 patients [12,29]. It is now understood that disease presentations may not include the classic findings and atypical clinical symptoms are frequently reported in individuals of non-Mediterranean origin. In Table 4 non-Mediterranean ancestry is not cause to rule out the diagnosis [30-38].
![]()
Ethnic Groups
MEFV genetic polymorphisms that alter pyrin function
Jordanians, Syrians, Egyptians, Moroccans, Tunisians, Iranians, Turks, North African Jews, Iraqi Jews
M694V
Jordanians, Egyptians, Algerians
M694I
Iranians, Turks
M680I
Jordanians, Egyptians, Israelis, Ashkenazi Jews
V726A
Ashkenazi Jews
E148Q
Table 4: Predominant MEFV genetic polymorphisms altering pyrin function in various Ethnic Groups. Familial Mediterranean Fever patient phenotype is variable and partially predictable based on genotype. Several polymorphisms in MEFV have been observed to occur at increased frequency in different ethnic groups. The M694V mutation is associated with the most severe, classic presentation of familial Mediterranean Fever. The V726A and E148Q mutations are associated with a less severe form of familial Mediterranean Fever. Adapted from Gershoni-Baruch R [23].
Listed in Table 5 are the predominant MEFV genetic polymorphisms altering pyrin function in various Ethnic Groups adapted from Gershoni-Baruch R, [23]. Several polymorphisms in MEFV occur at increased frequency in different ethnic groups. The M694V mutation is associated with the most severe, classic presentation of familial Mediterranean Fever. The V726A and E148Q mutations are associated with a less severe form of familial Mediterranean Fever.
![]()
Common Types of Cancer
Estimated New Cases 2013
Estimated Deaths 2013
All Cancer Sites
1,660,290
580,350
1.
Lung and Bronchus Cancer
228,190
159,480
2.
Colon and Rectum Cancer
142,820
50,830
3.
Breast Cancer
232,340
39,620
4.
Prostate Cancer
238,590
29,720
5.
Non-Hodgkin Lymphoma
69,740
19,020
6.
Bladder Cancer
72,570
15,210
7.
Kidney and Renal Pelvis Cancer
65,150
13,680
8.
Melanoma of the Skin
76,690
9,480
9.
Endometrial Cancer
49,560
8,190
10.
Thyroid Cancer
60,220
1,850
Table 5: Estimated number of new cancer cases and deaths in 2013 (SEER stat fact sheet).
Patients with atypical clinical presentation are prone to first experience symptoms at a later age (approximately 21.6 years), they are more likely to be misdiagnosed, and less likely to experience arthralgia. The most dire consequence of chronic untreated familial Mediterranean Fever is renal amyloidosis. Excessive release of the amyloid AA protein is a consequence of the hyperactive inflammatory response [28]; it is fully preventable with proper colchicine therapy [31-32]. In untreated individuals amyloidosis and subsequent renal disease typically occurs by age 40 [30]. In Mediterranean and non Mediterranean individuals, proper colchicine therapy dramatically reduces the incidence of febrile and serosal symptoms and also prevents the renal disease caused by amyloidosis [30-35,37,38].
The diagnostic tool with the highest specificity and sensitivity is analysis of the MEFV gene to detect disease-causing polymorphisms [23] Prior to the advent of the genetic testing for MEFV mutations, diagnosis of familial Mediterranean Fever was based on the response of the patient to prophylactic colchicine therapy [22]. Targeted molecular diagnostic testing is typically initiated by first screening for the most common polymorphisms with subsequent full gene sequencing if negative [24]. Diagnosis of familial Mediterranean Fever based on gene sequencing allows for determination of those whose clinical presentation does not match the classic definition of the disease [29]. Ashkenazi Jews with familial Mediterranean Fever are most frequently V726A homozygotes versus non- Ashkenazi Jews with familial Mediterranean Fever who tend to be M694V homozygotes. Compound heterozygosity is also frequently encountered in Ashkenazi Jews with familial Mediterranean Fever [37]. Patients of Ashkenazi Jewish heritage may be more prone to a familial Mediterranean Fever of mild clinical features relative to non-Ashkenazi Jews although it is reported that 3.5% of familial Mediterranean Fever Ashkenazi Jews develop renal failure without treatment [37]. Detection of the disease in these individuals is of the utmost importance as even mild renal failure is associated with increased mortality and decreased quality of life and may be clinically significant [37].
Autoinflammatory Syndromes
Familial Mediterranean Fever described in 1908 is one of three known monogenic periodic fevers and one of the twelve monogenic autoinflammatory syndromes comprehensively reviewed [39,40,43]. Autoinflammatory syndromes differ mechanistically from the more common and better characterized autoimmune disorders. Autoinflammation is a disorder of innate immunity whereas autoimmunity is a disorder of the adaptive immune system. In autoimmunity, the loss of one’s ability to distinguish self from non self is from B and T cell dysfunction and the effectors of inflammation are autoantibodies produced by B cells and by auto-reactive T lymphocytes. Autoinflammatory diseases are characterized by genetic polymorphisms in the mechanisms which initiate and control inflammation [41-43].
Inflammasome is a multiprotein intracytoplasmic scaffold complex that synthesizes the biologically active interleukin ( IL1). Inflammasome is the cell crucial to the regulation of innate immunity: its proper assembly allows for regular activation of caspase1 and physiological production of proinflammatory cytokines. Interleukin (IL 1) is the prototypic master cytokine affecting nearly all cell types. The erroneous assembly of the inflammasome leads to an exaggerated conversion of pro IL 1 beta to its active form and subsequent disproportionate overwhelming inflammatory response arising without provocation or attack by auto-antibodies or dysfunctional cytotoxic lymphocytes [43-46]. For the various monogenic inflammatory syndromes, the inflammasome represents an ideal point of convergence of most of these diseases. The classic familial Mediterranean Fever phenotype is an autosomal recessive condition. Familial Mediterranean Fever is caused by nonsense or missense mutations on chromosome 16p13.3 in the MEFV gene,which codes for the pyrin protein [45]. The MEFV gene is expressed in myeloid cells, and its expression is stimulated by tumor necrosis factor and interferon gamma. Pyrin is a 781 amino acid expressed in the cytoplasm of mature monocytes and neutrophils; it is detected in spleen lung and muscle [46]. In the inflammatory process, pyrin is an intranuclear regulator of the transcription of inflammatory peptides and the absence of pyrin function leads to unregulated production of IL-1 beta and oversecretion of inflammatory cytokines within neutrophils [47,48,49]. A partially predictable relationship exists between specific MEFV genotypes and the timing and severity of the disease, frequently quantified by the degree of associated renal amyloidosis. M694V homozygotes generally have the most severe clinical phenotype when compared to other characterized homozygous mutations as well as various compound heterozygote allele combinations [50].
Furthermore, the particular genotypes characterized and the associated haplotypes are differentially assorted between cultural and ethnic groups suggesting a predictable disease severity and course given a particular patient’s ethnicity and severity of the particular pyrin mutation [50,51]. Acute intermittent attacks in familial Mediterranean Fever develop because of neutrophil activation at the serosal surface. Serum amyloid AA deposition represents a late complication of untreated familial Mediterranean Fever. The precise trigger of the inflammation has been associated with numerous behavioral and physiological phenomena including strenuous exercise and cyclic hormone withdrawal in females. These inflammatory episodes subsequently cause the associated febrile symptoms [38].
Conclusion
Identification of genes involved in the modulation of inflammatory and apoptotic processes have improved our understanding of mechanisms linked to the aberrant activation of inflammasomes. The study case presented in this article illustrates the importance of recognizing a biomarker specific to the diagnosis of diseases that share common symptoms. As our approach to diagnostics grows increasingly sophisticated and molecular tools become more commonplace, it is likely that genotype-phenotype correlations will become more frequent and immediately clinically useful. Alterations in the approach to diagnostics with less rigid distinctions may allow for earlier treatment and improved quality of life for diseases whose symptoms fall ambiguously in the domain of the rheumatologist, the infectious disease specialist or the hematologist.
Acknowledgments
The research is supported by Elsa D and Carl E Hematology Endowment, Michigan State University. We appreciate the assistance of Kayla Bishop, Howard University Biology Major NSCS Scholar in preparing the manuscript and literature search.
References
- Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol. 2011; 7: 416-26.
- Hahn YS, Kim JG. Pathogenesis and clinical manifestations of juvenile rheumatoid arthritis. Korean J Pediatr. 2010; 53: 921-30.
- Prakken B1, Albani S, Martini A . Juvenile idiopathic arthritis. Lancet. 2011; 377: 2138-2149.
- Buoncompagni A, Barbano GC, Pistoia V, Fasce L, Micalizzi C. Childhood systemic lupus erythematosus: a review of 30 cases. Clin Exp Rheumatol. 1991; 9: 425-430.
- Hedrich CM, Zappel H, Straub S, Laass MW, Wieczorek K,et al. Early onset systemic lupus erythematosus: differential diagnoses, clinical presentation, and treatment options. Clin Rheumatol. 2011; 30: 275-83.
- Steere AC, Bartenhagen NH, Craft JE, Hutchinson GJ, Newman JH . The early clinical manifestations of Lyme disease. Ann Intern Med. 1983; 99: 76-82.
- Vasudevan B, Chatterjee M . Lyme borreliosis and skin. Indian J Dermatol. 2013; 58: 167-174.
- Cruz AT1, Starke JR . Clinical manifestations of tuberculosis in children. Paediatr Respir Rev. 2007; 8: 107-117.
- Whipple DV, Dunham EC. Congenital syphilis: Part l. Incidence, transmission, and diagnosis. J. Pediatr. 1938; 12: 386-398.
- Dorfman DH, Glaser JH. Congenital syphilis presenting in infants after the newborn period. N Engl J Med. 1990; 323: 1299-1302.
- Herremans T1, Kortbeek L, Notermans DW . A review of diagnostic tests for congenital syphilis in newborns. Eur J Clin Microbiol Infect Dis. 2010; 29: 495-501.
- Caso F, Rigante D, Vitale A, Lucherini OM, Costa L, et al. Monogenic autoinflammatory syndromes: state of the art on genetic, clinical, and therapeutic issues. International Journal of Hematology 2013; 2013: 1-15.
- Lamkanfi M, Dixit VM. Inflammasomes: guardians of cytosolic sanctity. Immunol Rev. 2009; 227: 95-105.
- Firestein GS, Budd RC, Gabriel SE, McInnes IB, O’Dell JR. Kelley’s textbook of Rheumatology. Philadelphia: W.B. Saunders Company; 2013.
- Padeh S. Periodic fever syndromes. Pediatr Clin North Am. 2005; 52: 577-609, vii.
- Sidoroff M, Karikoski R, Raivio T, Savilahti E, Kolho KL. High-sensitivity C-reactive protein in paediatric inflammatory bowel disease. World J Gastroenterol. 2010; 16: 2901-2906.
- Portincasa P, Scaccianoce G, Palasciano G. Familial mediterranean fever: a fascinating model of inherited autoinflammatory disorder. Eur J Clin Invest. 2013; 43: 1314-1327.
- Onen F. Familial Mediterranean fever. Rheumatol Int. 2006; 26: 489-496.
- Ozçakar ZB, Yalçinkaya F, Yüksel S, Ekim M. The expanded clinical spectrum of familial Mediterranean fever. Clin Rheumatol. 2007; 26: 1557-1560.
- Shohat M, Halpern GJ. Familial Mediterranean fever--a review. Genet Med. 2011; 13: 487-498.
- Zadeh N, Getzug T, Grody WW. Diagnosis and management of familial Mediterranean fever: integrating medical genetics in a dedicated interdisciplinary clinic. Genet Med. 2011; 13: 263-269.
- Shohat M, Halpern GJ. Familial Mediterranean fever--a review. Genet Med. 2011; 13: 487-498.
- Gershoni-Baruch R, Brik R, Shinawi M, Livneh A. The differential contribution of MEFV mutant alleles to the clinical profile of familial Mediterranean fever. Eur J Hum Genet. 2002; 10: 145-149.
- Caglayan AO, Demiryilmaz F, Ozyazgan I, Gumus H. MEFV gene compound heterozygous mutations in familial Mediterranean fever phenotype: a retrospective clinical and molecular study. Nephrol Dial Transplant. 2010; 25: 2520-2523.
- Eisenstein EM, Berkun Y, Ben-Chetrit E. Familial Mediterranean fever: a critical digest of the 2012-2013 literature. Clin Exp Rheumatol. 2013; 31: 103-107.
- Mimouni A, Magal N, Stoffman N, Shohat T, Minasian A. Familial Mediterranean fever: effects of genotype and ethnicity on inflammatory attacks and amyloidosis. Pediatrics. 2000; 105: E70.
- Lidar M, Livneh A. Familial Mediterranean fever: clinical, molecular and management advancements. Neth J Med. 2007; 65: 318-324.
- Lane T, Loeffler JM, Rowczenio DM, Gilbertson JA, Bybee A. AA amyloidosis complicating the hereditary periodic fever syndromes. Arthritis Rheum. 2013; 65: 1116-1121.
- Ben-Chetrit E, Touitou I. Familial mediterranean Fever in the world. Arthritis Rheum. 2009; 61: 1447-1453.
- L Obici, Merlini G. Amyloidosis in autoinflammatory syndromes. Autoimmun Rev. 2012; 12: 14-7.
- Rigante D, La Torraca I, Avallone L, Pugliese AL, Gaspari S, Stabile A. The pharmacologic basis of treatment with colchicine in children with familial Mediterranean fever. Eur Rev Med Pharmacol Sci. 2006; 10: 173-8.
- Ter HN, Lachmann H, Ozen S, Woo P, Uziel Y, et al. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis. 2013; 72: 678-85.
- Grateau G, Duruoz MT. Autoinflammatory conditions: when to suspect? How to treat? Best Pract Res Clin Rheumatol. 2010; 24: 401-411.
- Cantarini L, Capecchi PL, Lucherini OM, Laghi Pasini F,Galeazzi M. Familial Mediterranean fever diagnosed in an elderly patient. Clin Exp Rheumatol. 2010; 28: 91.
- Sayarlioglu M, Cefle A, Inanc M, Kamali S, Dalkilic E, et al. Characteristics of patients with adult-onset familial Mediterranean fever in Turkey: analysis of 401 cases. Int J Clin Pract. 2005; 59: 202-5.
- Gattorno M, Federici S, Pelagatti MA, Caorsi R, Brisca G. Diagnosis and management of autoinflammatory diseases in childhood. J Clin Immunol. 2008; 28 Suppl 1: S73-83.
- Lidar M, Kedem R, Berkun Y, Langevitz P, Livneh A. Familial Mediterranean fever in Ashkenazi Jews: the mild end of the clinical spectrum. J Rheumatol. 2010; 37: 422-425.
- Almeida de Jesus A, Goldbach-Mansky R . Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin Immunol. 2013; 147: 155-174.
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997; 17: 25-31.
- Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell. 1997; 90: 797-807.
- Meiorin SM, Espada G, Rosč C. Autoinflammatory diseases in pediatrics. Arch Argent Pediatr. 2013; 111: 237-243.
- Caso F, Rigante D, Vitale A, Lucherini OM, Costa L. Monogenic autoinflammatory syndromes: state of the art on genetic, clinical, and therapeutic issues. Int J Rheumatol. 2013; 2013: 513782.
- Rigante D, Frediani B, Galeazzi M, Cantarini L. From the Mediterranean to the sea of Japan: the transcontinental odyssey of autoinflammatory diseases. Biomed Res Int. 2013; 2013: 485103.
- Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012; 28: 137-161.
- Milhavet F, Cuisset L, Hoffman HM, Slim R, El-Shanti H. The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat. 2008; 29: 803-808.
- Chae JJ, Wood G, Masters SL, Richard K, Park G. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A. 2006; 103: 9982-9987.
- Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1β activation and severe autoinflammation in mice. Immunity. 2011; 34: 755-768.
- Stoffels M, Szperl A, Simon A, Netea MG, Plantinga TS. MEFV mutations affecting pyrin amino acid 577 cause autosomal dominant autoinflammatory disease. Ann Rheum Dis. 2014; 73: 455-461.
- Le HT, Harton JA. Pyrin- and CARD-only Proteins as Regulators of NLR Functions. Front Immunol. 2013; 4: 275.
- Cantarini L, Lucherini OM, Iacoponi F, Cimaz R, Simonini G. Development and preliminary validation of a diagnostic score for identifying patients affected with adult-onset autoinflammatory disorders. Int J Immunopathol Pharmacol. 2010; 23: 1133-1141.
- Cantarini L, Iacoponi F, Lucherini OM, Obici L, Brizi MG. Validation of a diagnostic score for the diagnosis of autoinflammatory diseases in adults. Int J Immunopathol Pharmacol. 2011; 24: 695-702.