
Review Article
Austin J Clin Med. 2014;1(4): 1016.
Genetics and Epigenetics in Tumorigenesis: Acting Separately or Linked?
Zhu X* and Wetta H
Department of Environmental Health, University of Cincinnati College of Medicine, USA
*Corresponding author: Xuegong Zhu, Department of Environmental Health, University of Cincinnati College of Medicine, Kettering Laboratory, 3223 Eden Avenue, Cincinnati, OH 45267-0056, USA
Received: May 31, 2014; Accepted: August 04, 2014; Published: August 11, 2014
Abstract
Tumorigenesis represents both genetic and epigenetic alterations. Genetic changes are defined as genomic DNA sequence changes, including point mutation, single nucleotide polymorphism (SNP) or copy number variability (CNV), including loss of heterozygozity (LOH), gain copy number or deletion. On the other hand, epigenetic modification is responsible for regulation of gene expression without changing the underlying DNA sequence. Such mechanisms include DNA methylation, post-translational modifications of the histone proteins, and micro RNA, resulting in selective gene activation and/or in activation. As opposed to solely isolated genetic and epigenetic components acting in tumorigenesis, the complex nature of cancer seems to stem from the interaction of these mechanisms.
Keywords: Genetics; Epigenetics; Tumor
Tumorigenesis
Genes associated with disease susceptibility can contribute to the onset of cancer and other diseases through multiple mechanisms. As shown in Figure 1, there are two copies, or alleles, of every gene, which are located on two separate chromosomes: one from the mother and one from the father. If a gene is an oncogene, an active mutation or epigenetic activation of only one gene copy is enough to drive expression of the gene. For example, MAGE-A3 is not expressed in normal pituitary tissue; however it is activated in pituitary tumors through promoter DNA de-methylation [1].
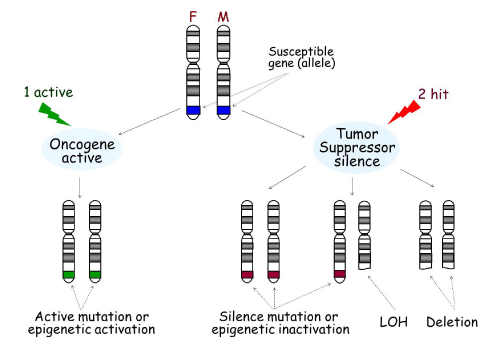
Tumorigenesis.
Development of cancer can occur through two main mechanisms. One “active,„ usually an active mutation or epigenetic activation in oncogenes in either the father or mother (seen in green), is necessary for the activation of the oncogene, leading to tumorigenesis. On the other hand, tumor suppressors must be silenced by two “hits,„ including silence mutation, epigenetic inactivation, Loss of Heterozygosity (LOH), or deletion (seen in red). F: Father; M: Mother.
Figure 1:Tumorigenesis.
Development of cancer can occur through two main mechanisms. One “active,„ usually an active mutation or epigenetic activation in oncogenes in either the father or mother (seen in green), is necessary for the activation of the oncogene, leading to tumorigenesis. On the other hand, tumor suppressors must be silenced by two “hits,” including silence mutation, epigenetic inactivation, Loss of Heterozygosity (LOH), or deletion (seen in red). F: Father; M: Mother.
If a gene is a tumor suppressor, it must be silenced by “two hits„. Mechanisms by which tumor suppressors are silenced include mutation, deletion, LOH or epigenetic alteration. At present, there are a multitude of tumor suppressor genes that have been shown to be inactivated this way [2].
Tumorigenesis Represents both Genetic and Epigenetic Alterations
Both genetic and epigenetic changes can lead to the development of cancer (Figure 2). Genetic alterations, changes to the underlying DNA sequence of genes, transpire via somatic mutation, single nucleotide polymorphisms (SNPs), and copy number variation (CNV), including LOH, deletion, and gain of copy number. Point mutation is genetic modification in any somatic cells in an individual, whereas SNPs are changes of a single nucleotide in certain individuals in a population. CNV results in an alteration of the number of copies of certain regions of the genome by more than 1,000 nucleotides, such as a loss of one copy (LOH), deletion of both copies, and a gain in copy number. Genetic modifications are relatively difficult to reverse.
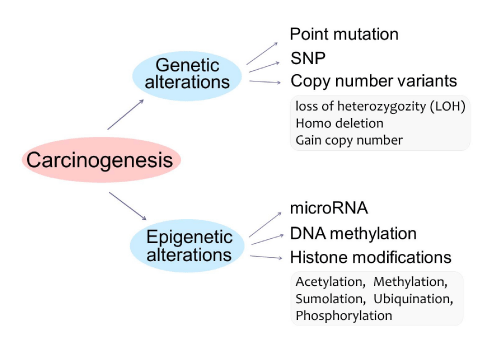
Figure 2: Tumorigenesis represents both genetic and epigenetic alterations. Genetic alterations include point mutation, single nucleotide polymorphisms (SNPs), or copy number variability (CNV), including LOH, deletion, and gain of copy number. Epigenetic alterations include: micro RNA, DNA methylation, or histone modifications, including Acetylation, Methylation, Sumolation, Ubiquination, and Phosphorylation.
Epigenetic alterations include micro RNA (mi RNA), small non-coding RNA molecules that bind to their complementary sequence, thus preventing transcription of a gene; DNA methylation, the addition of methyl groups to specific nucleotides of a sequence, preventing expression of a gene; and histone modifications, the alteration of accessibility to the DNA sequence. Many drugs can influence gene expression by inhibiting the activity of enzymes involved in epigenetic regulation, providing numerous potential therapies for disease.
Genetics in Tumorigenesis
Point Mutations
Point mutations are changes that alter only one or a few nucleotides along the nucleotide sequence of the genome, resulting in unrepaired damage to DNA. Point mutations in multiple tumor suppressor genes have been shown to cause cancer. For instance, point mutations in adenomatous polyposis coli (APC) promote tumorigenesis [3].
Additionally, the change of an oncogene from normal to its cancerous form can also be caused by a simple point mutation in the sequence of the gene. For example, a change in the ras oncogene, located on human chromosome 11, from guanine to cytosine is frequently associated with bladder cancer [4].
Single nucleotide polymorphisms (SNPs)
SNPs are specific sites within a human genome at which some individuals will have one nucleotide present while other individuals will have a different one. SNPs are a hot topic due to their recently discovered involvement in disease diagnosis, drug resistance, and patient outcome. SNPs may influence gene expression or gene function in two ways (Figure 3). If the SNP is located in an intron, it may change the binding ability of transcription factors to the SNP area, consequently influencing gene expression. This is seen in SNPs located in fibroblast growth factor receptor 2 (FGFR2) intron 2 [5].Conversely, if the SNP is located in the exon coding region it may affect the structure of the translated protein, which may affect its sensitivity to upstream signal or effectiveness in downstream function. This is the case for fibroblast growth factor 4 (FGFR4) SNPs in the trans membrane domain encoding 388Gly/Arg [6].
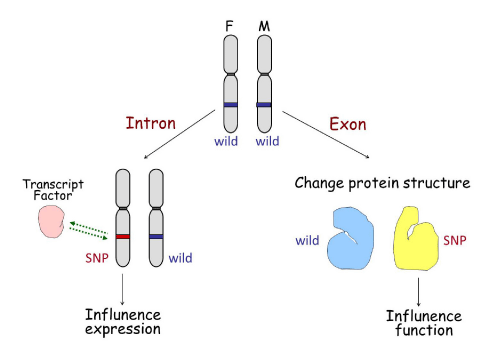
SNP influence on gene expression or function.
The left half shows a SNP located in an intron of a gene, which may change the binding ability of transcription factors to the area of the SNP, influencing gene expression accordingly. The right half represents a SNP located in an exon of a gene that may affect the transcription and translational codes, altering the structure of the translated protein, and therefore affecting its sensitivity to upstream signals or its effectiveness in downstream function.
Figure 3:SNP influence on gene expression or function.
The left half shows a SNP located in an intron of a gene, which may change the binding ability of transcription factors to the area of the SNP, influencing gene expression accordingly. The right half represents a SNP located in an exon of a gene that may affect the transcription and translational codes, altering the structure of the translated protein, and therefore affecting its sensitivity to upstream signals or its effectiveness in downstream function.
Loss of heterozygosity
Human somatic cells contain two copies of the genome, one from each parent. LOH occurs when a chromosomal region is lost from one chromosome, leaving just one copy of the genes within the region. This condition can indicate the absence of functional tumor suppressor genes encoded in the lost chromosomal region. LOH is often associated with cancerous phenotypes and tumor progression. For example, gynecological carcinosarcoma has both sarcomatous and carcinomatous components. LOH examination revealed that the two components shared the same LOH pattern, indicating that they arose from a single malignant epithelial precursor, strongly supporting that most gynecological carcinosarcomas have a monoclonal origin [7].
Epigenetics in Tumorigenesis
DNA methylation
Tumor suppressor genes can be suppressed by DNA methylation
Most DNA methylation occurs at the cytosine in CpG dinucleotide sites. The majority of these sites are concentrated in CpG islands, which are typically located in the promoter region and first exon of genes [8]. Hypermethylated CpG islands can recruit Methyl- CpG-binding Protein (MeCP), which block transcription factors from binding to the promoter region, resulting in abrogated gene expression (Figure 4). MeCP can also recruit Histone Deacetylase (HDAC), which drives chromatin remodeling through histone methylation. This inhibits transcription factors from binding to the promoter region. DNA methylation can be reversed by inhibiting the activity of the methylation control enzyme DNMT, causing re-activation of gene expression [9].
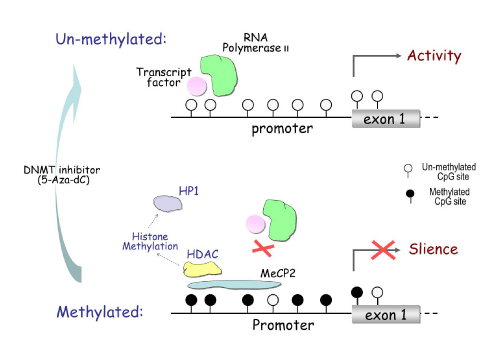
DNA methylation inhibits gene transcription.
The upper half shows the unmethylated condition, where open circles represent unmethylated CpG sites. Unmethylated CpG sites in the promoter region of a gene allow for the binding of transcription factors and RNA polymerase to activate gene transcription. Seen below, hyper methylation at CpG sites recruits Methyl-CpG-binding Protein 2 (MeCP2), which blocks transcription factors from binding to the promoter and thus represses transcription. The MeCP2 can also recruit Histone Deacetylase (HDAC), causing histone methylation, which can further recruit Heterochromatin Protein 1 (HP1), triggering chromatin remodeling.
Figure 4:DNA methylation inhibits gene transcription.
The upper half shows the unmethylated condition, where open circles represent unmethylated CpG sites. Unmethylated CpG sites in the promoter region of a gene allow for the binding of transcription factors and RNA polymerase to activate gene transcription. Seen below, hyper methylation at CpG sites recruits Methyl-CpG-binding Protein 2 (MeCP2), which blocks transcription factors from binding to the promoter and thus represses transcription. The MeCP2 can also recruit Histone Deacetylase (HDAC), causing histone methylation, which can further recruit Heterochromatin Protein 1 (HP1), triggering chromatin remodeling.
At present, there are numerous known tumor suppressor genes silenced through DNA methylation in tumors. The mismatch repair gene, hMLH1, is silenced in colon, gastric, and endometrial tumors; without hMLH1, cells cannot repair mismatch mutations in DNA, disrupting the proper functioning of many other proteins whose transcription is regulated by hMLH1. Other genes involved in maintaining cellular stability, such as cell cycle regulators, (p16INK4a), DNA repair (MGMT), apoptosis (DAPK), and cell adherence (CDH1) have also been shown to have hypermethylated promoter regions in tumors, leading to their subsequent silencing [10].
Oncogenes can be activated by DNA de-methylation
In addition to the inactivation of tumor suppressor genes, DNA demethylation and subsequent activation of oncogenes can also result in the development of cancer [11]. Melanoma-associated antigen A, of the MAGE family, exhibits the oncogenic mechanism of tumorigenesis. While MAGE-A genes are not normally expressed in tissues other than test is and placenta, DNA demethylation allows for transcription of the MAGE-A genes in multiple types of human cancers [12].
Histone modification
Histone modifications can affect local chromatin structure that contributes to the determination of whether a gene is transcribed or repressed. Nucleosomes, which are the fundamental repeating subunits of chromosomes, consist of approximately 146 base pairs of DNA and four pairs of histones: H2A, H2B, H3 and H4. Acetylation of lysine residues on histones H3 and H4 lead to the formation of an open chromatin structure (euchromatin) that permits access to DNA by regulatory proteins such as transcription factors. Conversely, methylation of lysine 9, 27, or other residues on H3 leads to the formation of a closed structure (heterochromatin), which blocks transcription factor binding to DNA and facilitates transcriptional repression (Figure 5). Histone modification is regulated by the enzymes histone acetyltransferase (HAT), histone deacetylase (HDAC), histone methyltransferase (HMT), and histone demethylase (HDM). Drugs such as the HDAC inhibitor trichostatin A (TSA) can increase histone acetylation, thereby inducing gene expression [13].
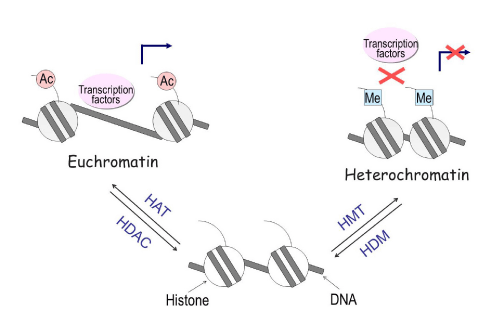
Figure 5: Histone modifications influence gene expression through chromatin remodeling. Acetylation of histones leads to the formation of a loose condition of chromatin (euchromatin), permitting access to DNA by transcription factors thus allowing for genes to be active. Conversely, methylation of histones leads to the formation of a condensed condition of chromatin (heterochromatin), a closed structure that blocks transcription factors from binding to DNA, facilitating transcriptional repression. The enzymes Histone Acetyl transferase (HAT), Histone deacetylase (HDAC), Histone methyl transferase (HMT), and Histone demethylase (HDM) regulate the process of histone modification.
DNA methylation and histone modification function independently or together to regulate gene expression
DNA methylation can directly suppress gene expression
Estrogen Receptor Beta (ER-β) is dynamically regulated by DNA methylation in prostate cancer throughout its progression. During the development of prostate cancer in the gland, diminished expression of ER-β occurs through methylation of the CpG is land at the promoter region of the gene, thereby leading to its transcriptional inactivation. As the cancer metastasizes in bone and lymph nodes, ER-β reappears by demethylation. Evidence obtained through methylated oligonucleotides, which can mediate sequence-specific methylation in cellulo, specifically methylate the promoter region of a gene, revealing that methylation can directly suppress ER-β expression [14].
Histone modification can independently regulate gene expression
Interestingly, genes like DNMT3b, which have large CpG island in its promoter, are actually only regulated at the histone level. DNMT3b and other DNA methyl transferases maintain DNA methylation patterns of the entire genome. Methylation of CpG islands using bisulfite sequencing and combined bisulfite restriction analysis (COBRA) reveals that DNA methylation in significantly contributes to the regulation of DNMT3b expression, as compared to anti-methyl-histone H3-Lys9 and AcH3 antibodies used for acetylation and demethylation of the histone mark. The tests show that histone modifications are more relevant in regulating the expression of DNMT3b [15].
DNA methylation and histone modification can co-regulate gene expression
In pituitary tumors, Ikaros, a transcription factor especially pertinent in the immune and endocrine systems, and FGFR2,a membrane-bound protein present in blood vessels and secreted by adipocytes, are regulated by both DNA and histone modification. The jointly altered expression of the genes is evident through the use of two methods. One method treats cells with a DNA methyltransferase inhibitor to reveal the effects of DNA methylation, while treatment with a HDAC inhibitor allows for the investigation of alterations in expression by histone modification. Both treatments result in up-regulated protein levels, exposing a joint regulation of the gene by both DNA methylation and histone modification. A second method utilizes bisulfite sequencing and chromatin immune precipitation (ChIP) to examine specific regions of a gene which are targets of DNA methylation and histone modification, respectively. These techniques also support that a combination of DNA methylation and histone modification is involved in the regulation of these genes [16,17].
Epigenetic alteration is not only responsible for single gene regulation
Epigenetic alteration is not only responsible for single gene regulation, but also plays an important role in gene-gene interaction and signal transduction. For example, FGFR2b and Estrogen regulate MAGE-A3, which then regulates p53 and p21 in pituitary tumors. At each level, this regulation is occurring through histone modification [1] (Figure 6).Ultimately, epigenetic regulation provides numerous potential therapies for disease by inhibiting the activity of enzymes involved.
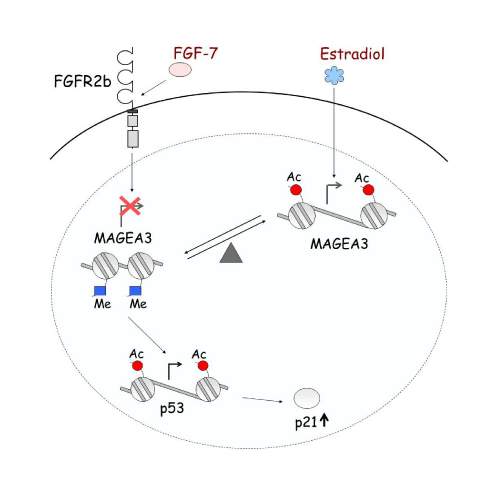
Epigenetic regulation network.
FGF7/FGFR2-IIIb and estradiol oppositely regulate the expression of the MAGE-A3 oncogene in pituitary tumors. The two systems create a balance regulating MAGE-A3 expression through histone acetylation and methylation. Estradiol increases acetylation level, enhancing transcription of MAGE-A3. Down-regulation of MAGE-A3 by FGF7/FGFR2-IIIb increases acetylation of p53, resulting in increased p53 expression, and consequently induction of p21.
Figure 6:Epigenetic regulation network.
FGF7/FGFR2-IIIb and estradiol oppositely regulate the expression of the MAGE-A3 oncogene in pituitary tumors. The two systems create a balance regulating MAGE-A3 expression through histone acetylation and methylation. Estradiol increases acetylation level, enhancing transcription of MAGE-A3. Down-regulation of MAGE-A3 by FGF7/FGFR2-IIIb increases acetylation of p53, resulting in increased p53 expression, and consequently induction of p21.
Genetic and Epigenetic Mechanisms function together in tumorigenesis
LOH and DNA methylation
On top of isolated epigenetic alterations, genetic sequence changes can simultaneously contribute to cancer and other diseases. For example, FGFR2, which allows fibroblast growth factors (FGFs) to transmit signals involved in cell differentiation and proliferation, is down-regulated by both epigenetic and genetic mechanisms in breast cancer. LOH and DNA methylation in specific regions of FGFR2 determine breast cancer progression by loss of the gene or by limiting transcriptional access [18].
Additional genes in which LOH and DNA methylation function together are in breast cancer 1 (BRCA1) and fragile histidine triad (FHIT). The famous tumor suppressor gene, BRCA1, is silenced in many cases of primary breast and ovarian carcinomas, by a combination of promoter hyper methylation and LOH [19] FHIT is another tumor suppressor gene inactivated by LOH and methylation of 5-CpG [20].
SNPs and epigenetics
FGFR2 is also regulated by both SNPs and histone modification. SNPs in FGFR2 intron 2 can recruit active proteins to increase FGFR2 expression, and histone acetylation can modulate these selected SNPs in generating variable FGFR2 levels in breast cancer [5]. In FGFR4, a SNP changing a glycine to an arginine is present at amino acid position 388 in the Trans membrane domain. Using micro dissected primary breast cancer specimens, it was shown that DNA methylation is mainly found in the wild type388 glycine allele, a probable tumor suppressor. In addition, DNA methylation at the FGFR4 wild type 388 allele was confirmed to be related to breast cancer progression by mouse xenograft model [6].
DNA methylation and genetic mutations
According to the clustering analyses of genetic and epigenetic abnormalities, Shen, et al identified three distinct DNA methylation groups of colon cancers, CpG island methylator phenotype CIMP1, CIMP2, and CIMP negative. Genetically, these three groups correspond to very distinct profiles. CIMP1 is characterized by microsatellite instability (MSI) and BRAF mutations, along with rare KRAS or p53 mutations. CIMP2 is associated with KRAS mutations and rare MSI, BRAF, or p53 mutations. CIMP-negative cases have a high rate of p53 mutations and lower rates of MSI or mutations of BRAF or KRAS [21].
Micro RNAs, SNP and DNA Methylation
Genetic association analysis of 15 mi RNA genes found that a common SNP (rs11614913, C→T) in hsa-miR-196a-2 is considerably associated with decreased breast cancer risk, as is hyper methylation of Cp Gisland upstream of the miR-196a-2 precursor. Analysis of mutagenesis showed that the variant miR-196a-2 may have a potentially oncogenic role in breast cancer because of its diminished processing of the mi RNA precursor to maturity and its decreased ability to regulate the mi RNA’s target genes [22].
Conclusion
As the field of molecular genetics advances, it is clear that cancer is a multi faceted disease, gaining its strength from its complex nature. To fully comprehend the cause and course of this modestly understood disease, scientists must look at cancer development with an inter-mechanistic approach. Multiple systems of abnormality work together in cancer; therefore research should focus on the interaction between genetics and epigenetics, rather than solely on a distinct aspect of either. The same idea applies to clinical practice, in that cancer treatments often target one characteristic defect. True progress in the field will be achieved when both researchers and clinicians can use a genome-wide tactic to treat, and cure, cancer.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (Grant 81172519).
References
- Zhu X, Asa SL, Ezzat S. Fibroblast growth factor 2 and estrogen control the balance of histone 3 modifications targeting MAGE-A3 in pituitary neoplasia. Clin Cancer Res. 2008; 14: 1984-1996.
- Lamlum H, Ilyas M, Rowan A, Clark S, Johnson V, Bell J, et al. The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: a new facet to Knudson's 'two-hit' hypothesis. Nat Med. 1999; 5: 1071-1075.
- Minde DP, Anvarian Z, Rüdiger SG, Maurice MM. Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer? Mol Cancer. 2011; 10: 101.
- Knowles MA, Williamson M. Mutation of H-ras is infrequent in bladder cancer: Confirmation by single-strand conformation polymorphism analysis, designed restriction fragment length polymorphisms, and direct sequencing. Cancer Res. 1993; 53: 133-139.
- Zhu X, Asa SL, Ezzat S. Histone-acetylated control of fibroblast growth factor receptor 2 intron 2 polymorphisms and isoform splicing in breast cancer. Mol Endocrinol. 2009; 23: 1397-1405.
- Zhu X, Asa SL, Ezzat S. Loss of Heterozygosity and DNA Methylation Affect Germline Fibroblast Growth Factor Receptor 4 Polymorphism to Direct Allelic Selection in Breast Cancer. Am J Pathol. 2010; 177: 2860-2869.
- Fujii H, Yoshida M, Zhu X, Matsumoto T, Hamano Y, Fukunaga M, et al. Frequent genetic heterogeneity in the clonal evolution of gynecological carcinosarcoma and its influence on phenotypic diversity. Cancer Res. 2000; 60: 114-120.
- Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007; 8: 286-298.
- Nazemalhosseini Mojarad E, Kuppen PJ, Aghdaei HA, Zali MR. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol Hepatol Bed Bench. 2013; 6: 120-128.
- Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002; 21: 5427-5440.
- Byler S, Goldgar S, Heerboth S, Leary M, Housman G, Moulton K, et al. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. 2014; 34: 1071-1077.
- Wischnewski F, Pantel K, Schwarzenbach H. Promoter demethylation and histone acetylation mediate gene expression of MAGE-A, -A2, -A3, and -A12 in human cancer cells. Mol Cancer Res. 2006; 4: 339-349.
- Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet. 2001; 10: 687-692.
- Zhu X, Leav I, Leung YK, Wu M, Liu Q, Gao Y, et al. Dynamic regulation of estrogen receptor-beta expression by DNA methylation during prostate cancer development and metastasis. Am J Pathol. 2004; 164: 2003-2012.
- Zhu X, Mao X, Hurren R, Schimmer A, Ezzat S, Asa S. Deoxyribonucleic acid methyltransferase 3B promotes epigenetic silencing through histone 3 chromatin modifications in pituitary cells. J ClinEndocrinolMetab. 2008; 93: 3610-3617.
- Zhu X, Asa SL, Ezzat S. Ikaros is regulated through multiple histone modifications and deoxyribonucleic acid methylation in the pituitary. Mol Endocrinol. 2007; 21: 1205-1215.
- Zhu X, Lee K, Asa SL, Ezzat S. Epigenetic silencing through DNA and histone methylation of fibroblast growth factor receptor 2 in neoplastic pituitary cells. Am J Pathol. 2007; 170: 1618-1628.
- Zhu X, Asa SL, Ezzat S. Genetic and epigenetic mechanisms down-regulate FGF receptor 2 to induce melanoma-associated antigen A in breast cancer. Am J Pathol. 2010; 176: 2333-2343.
- Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000; 92: 564-569.
- Yang Q, Nakamura M, Nakamura Y, Yoshimura G, Suzuma T, Umemura T, et al. Two-hit inactivation of FHIT by loss of heterozygosity and hypermethylation in breast cancer. Clin Cancer Res. 2002; 8: 2890-2893.
- Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A. 2007; 104: 18654-18659.
- Hoffman AE, Zheng T, Yi C, Leaderer D, Weidhaas J, Slack F, et al. microRNA miR-196a-2 and breast cancer: a genetic and epigenetic association study and functional analysis. Cancer Res. 2009; 69: 5970-5977.