
Mini Review
Austin J Clin Neurol 2015;2(3): 1033.
Vitamin D, Cell Signalling Phenotypic Stability and Alzheimer’s Disease
Berridge MJ*
Laboratory of Molecular Signalling, Babraham Research Campus, UK
*Corresponding author: Berridge MJ, Laboratory of Molecular Signalling, the Babraham Institute, Babraham Research Campus, Cambridge CB22 3AT, UK
Received: March 04, 2015; Accepted: March 25, 2015; Published: April 07, 2015
Abstract
Vitamin D deficiency has been linked to many human diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Multiple Sclerosis (MS), hypertension and cardiovascular disease. A Vitamin D phenotypic stability hypothesis, which is developed in this review, attempts to describe how this vital hormone acts to maintain healthy cellular functions. This role of Vitamin D as a guardian of phenotypic stability seems to depend on its ability to maintain the redox and Ca2+ signalling systems. It is argued that its primary action is to maintain the expression of those signalling components responsible for stabilizing the low resting state of these two signalling pathways. This phenotypic stability role is facilitated through the ability of vitamin D to increase the expression of both Nrf2 and the anti-aging protein Klotho, which are also major regulators of Ca2+ and redox signalling. A decline in Vitamin D levels will lead to a decline in the stability of this regulatory signalling network and may account for why so many of the major diseases in man, which have been linked to vitamin D deficiency, are associated with a dysregulation in both ROS and Ca2+ signalling as is described for Alzheimer’s disease (AD).
Keywords: Vitamin D; Calcium; Klotho; Nrf2; Alzheimer’s disease
Introduction
Vitamin D deficiency is a major human epidemic [1]. There is increasing evidence for a link between Vitamin D deficiency and many of the major human diseases (Figure 1). While the evidence for such associations is strong, particularly in the case of neural diseases such as Alzheimer’s disease, Parkinson’s disease and Multiple Sclerosis (MS), there is little information as to why a deficiency in Vitamin D can have such serious consequences. In an attempt to answer this question, I have developed a Vitamin D phenotypic stability hypothesis that sets out to explain why Vitamin D is such an important hormone responsible for maintaining normal cellular functions [2].
Figure 1: Vitamin D deficiency has been linked to a large number of human
diseases.
Vitamin D - a custodian of phenotypic stability in cell signalling pathways
When cells differentiate during development, they select out those components of their signalling toolkits to assemble specific signalling systems such as the Ca2+ and redox signalling systems. For normal cell responses, it is essential that transcription of all the components that make up such Ca2+ and redox signalling phenotypes are maintained and it seems that Vitamin D, working together with Nrf2 and Klotho, is the major custodian of such phenotypic stability (Figure 2).
Vitamin D acts by binding to the Vitamin D Receptor (VDR), which interacts with the Retinoid X Receptor (RXR) before binding to the Vitamin D Response Element (VDRE), located on a large number of vitamin D-sensitive target genes. The VDR can also be partially activated by a number of other ligands, which have health benefits such as Lithocholate (LCA), curcumin, omega 3 fatty acids and resveratrol [3]. Vitamin D controls the expression of Nrf2 and the anti-aging protein Klotho, which are also important regulators of multiple cellular signalling systems (Figure 2). Many of the genes that are controlled by the Vitamin D/Klotho/Nrf2 regulatory network function to maintain Ca2+ and redox homeostasis. For example, Vitamin D increases the expression of Ca2+ pumps, exchangers and buffers to maintain low levels of Ca2+. Similarly, Vitamin D together with Klotho and Nrf2 all increase cellular antioxidants to maintain the normal reducing environment within the cell.
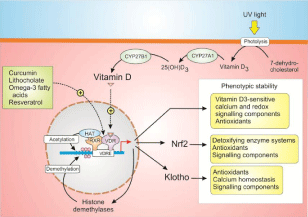
Figure 2: Vitamin D, which is formed through a series of reactions beginning
with UV-induced photolysis of 7-dehydrocholesterol to form Vitamin D3,
which then undergoes two hydroxylation reactions to form the active Vitamin
D. Vitamin D then acts on the Vitamin D Receptor (VDR) to increase the
expression of a large number of genes many of which regulate cell signalling
pathways so as to maintain phenotypic stability. Vitamin D also controls the
epigenetic landscape of its target genes by regulating both acetylation and
methylation.
Vitamin D regulation of the epigenetics landscape
In keeping with its proposed role in maintaining phenotypic stability, Vitamin D controls the epigenetic landscape of its multiple gene promoters to maintain the transcription activity of all the genes that operate in its regulatory network (Figure 2). Vitamin D controls both the acetylation and methylation states of its promotor regions. The VDR complex recruits histone acetylases such as p300/CBP and SRC-1 that acetylate chromatin and it increases the expression of a number of DNA demethylases. This ability of Vitamin D to modulate the epigenetic landscape may contribute to its ability to maintain phenotypic stability and to prevent the onset of multiple diseases [2].
Vitamin D deficiency and age-related decline in human cognition
There is an age-related decline in the ability of human skin to synthesize Vitamin D [4] and this may account for the decline in the level of Vitamin D and Klotho during aging. Vitamin D deficiency may thus contribute to the normal aging process through dysregulation of cell signalling pathways such as those operated by the Ca2+ and redox cell signalling pathways. Dysregulation of Ca2+ signalling, which is closely linked to mitochondrial dysfunction and ROS formation, has been implicated in aging [5]. Another example of an alteration in Ca2+ signalling during aging is the decline in the Ca2+ buffer cabining in neurons [6]. The decline in this buffer may also be linked to a deficiency in Vitamin D, which normally maintains the expression of calbindin. During aging, therefore a decline in Vitamin D may result in an elevation in the Ca2+ and redox signalling systems resulting in phenotypic instability and the onset of many age-related human diseases.
There is increasing evidence that a deficiency in Vitamin D can lead to a decline in neurological functions such as cognition and various neurodegenerative diseases [7]. Strong support for such a notion has come from the study of the decline in cognition in aging rats that is driven by a marked increase in the amplitude of the slow After Hyper Polarization (sAHP) that depend on a build-up of Ca2+ that activates the SK potassium channel [8] (Figure 3). This Ca2+ signal, which depends on the opening of L-type voltage-dependent Ca2+ channels that provides trigger Ca2+ to activate ryanodine receptors (RYRs), inhibits memory by curtailing the spiking activity necessary for LTP, whereas the increase in Ca2+ stimulates calcineurin to induce the Long-Term Depolarization (LTD) that erases memories [9]. The development of this sAHP during aging depends on dysregulation of both Ca2+ and ROS signalling that can be directly attributed to Vitamin D deficiency (Figure 3). The increase in ROS signalling sensitizes the RYRs and this can be reversed by treating neurons with Dithiothreitol (DTT) [10].

Figure 3: Age-related decline in cognition and its reversal by the Vitamin D/
Klotho/NRF2 regulatory network. In aged rats, the neuronal action potentials
activate L-type Ca2+ channels that provide trigger Ca2+ that stimulates
ryanodine receptors (RYRs) to generate a large Ca2+ signal that results in
memory loss through two processes. Firstly, the Ca2+ acts on SK channels
resulting in a slow After Hyper Polarization (sAHP) that causes spike failure
and a decrease in the Long-Term Potentiation (LTP) responsible for memory
formation. Secondly, the prolonged elevation in cytosolic Ca2+ activates
calcineurin that increases Long-Term Depression (LTD) resulting in memory
loss. The Vitamin D/Klotho/NRF2 regulatory network acts to suppress
the abnormal elevation in Ca2+ through a number of mechanisms that act
to reduce both Ca2+ and Reactive Oxygen Species (ROS) signalling as
described in the text.
The Vitamin D/Klotho/NRF2 regulatory system can prevent the dysregulation of Ca2+ and ROS signalling through multiple mechanisms (Figure 2). Vitamin D suppress the expression of the L-type Ca2+channel [11] that initiates the Ca2+ signal that induces the sAHP and it also maintains the expression of PMCA and NCX1, which extrude Ca2+ from the cell (Figure 3). Klotho acts to stimulate the Na+/K+-ATP sae responsible for maintaining the Na+ gradient necessary for Ca2+ extrusion by NCX1. Finally, NRF2 increases the expression of many antioxidants that ensure that ROS levels are kept low, which will prevent the sensitization of the RYRs that are triggering the sAHP and memory erasure.
The central role of vitamin D deficiency in this neuronal dysregulation and cognitive decline can be reversed by treating neurons with vitamin D that dramatically reduces the sAHP [12]. When tested on aging rats, Vitamin D was found to enhance hippocampal synaptic function and, more significantly, it could prevent the decline in cognition [13]. Such activation of the RYRs by L-type Ca2+ channels, which is responsible for this age-related decline in cognition, is a significant feature of Alzheimer’s disease (AD) (Figure 4) [14]. Such observations are consistent with the finding that a deficiency in Vitamin D predicts a decline in human cognition that occurs with aging [15-17]. The decline in the level of Klotho during aging, which is probably linked to the decline in Vitamin D, may also contribute to the decline in cognition. A single allele of the KL-VS variant of the Klotho gene, which greatly enhances longevity, markedly reduces the age-related decline in human cognition [18].
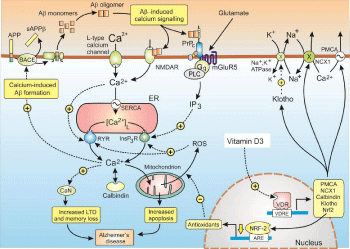
Figure 4: Dysregulation of Ca2+ signalling in Alzheimer’s disease. Hydrolysis
of the Amyloid Precursor Protein (APP) by β-site APP-Cleaving Enzyme
(BACE) generates the amyloid β (Aβ) monomers that then form the Aβ
oligomers that result in Aβ-induced calcium signalling. One of the main
actions of Aβ is to bind to the cellular prion protein (PrPC) that is coupled
to the mGluR5 receptor to generate inositol 1,4,5-trisphosphate (InsP3) to
increase the release of Ca2+ from the ER. Much of the released Ca2+ enters
the mitochondria to induce an increase in the formation of Reactive Oxygen
Species (ROS), which sets up a positive feedback loop because the ROS
acts to sensitize both the InsP3Rs and the Ryanodine Receptors (RYRs). The
increased level of Ca2+ then acts on calcineurin (CaN) that enhances memory
loss by increasing Long-Term Depression (LTD). Vitamin D counteracts this
abnormal elevation of both Ca2+ and ROS through a number of mechanisms
as described in the text.
Despite many attempts to explain aging, there is no consensus as to what the mechanism might be. These studies illustrating how Vitamin D deficiency contributes to the age-related decline in cognition suggests the interesting possibility that Vitamin D might be a key element in determining the rate of the aging process and the onset of age-related diseases such as Alzheimer’s disease (AD) as described below (Figure 3).
Vitamin D deficiency and Alzheimer’s disease (AD)
Why is it that some individuals develop Alzheimer’s disease (AD) as they grow older while others do not? The answer may lie in their relative Vitamin D levels that may determine the rate of aging as proposed above. Those with low Vitamin D levels will experience abnormal elevations in Ca2+, similar to those described in aged rats (as described above), that will trigger calcium-induced Aβ formation [19- 22] (Figure 4). Such Ca2+-induced increase in amyloidal formation then initiates a positive feedback loop because it is followed by Aβ- induced Ca2+ signalling [2]. Such a scenario may explain the sporadic nature of AD.
The Aβ can activate Ca2+ signalling through different mechanisms. It can bind to the cellular prion protein (PrPC), which is coupled to mGluR5 to increase the formation of InsP3 to release internal Ca2+ [23]. Aβ can also activate the NMDARs to increase Ca2+ entry. An increase in ROS formation that occurs in AD will also enhance Ca2+ signalling by sensitizing both the InsP3Rs and the RYRs (Figure 4). Such ROS-dependent dysregulation of Ca2+ release by neuronal RYRs, which is triggered by entry of Ca2+ through the L-type Ca2+ channels, is a feature of age-related memory loss as described earlier [24]. Such an interaction between the L-type Ca2+ channels and RYRs (Figure 4) is a significant feature of AD [14]. This persistent elevation in the resting level of Ca2+ may act to induce Long-Term Depressio n (LTD) to continuously erase memories shortly after they are formed during the wake period [25,26].
There are an increasing number of studies indicating that a deficiency in Vitamin D may contribute to the onset of AD [27- 29]. Since AD seem to be caused by abnormal elevations in Ca2+, the deleterious effect of vitamin D deficiency may be explained by a decrease in its normal role as a custodian of Ca2+ and ROS homeostasis. Vitamin D may act to reduce the onset of AD through its ability to stimulate the expression of Ca2+ pumps (PMCA and NCX1) and Ca2+ buffers such as calbindinand parvalbumin and to reduce the expression of the L-type Ca2+ channels. Neuronal levels of cabining are known to be reduced in AD [6,30]. The level of Nrf2, which is markedly reduced in the brain of patient with AD [31], is increased by Vitamin D. Cognition in AD transgenic mice was markedly improved following vector-mediated expression of Nrf2 in the hippocampus [32]. Nrf2 may act to reduce the symptoms of AD by maintaining the cellular level of the redox buffer GSH, which is a critical factor in preventing AD [33].
In a mouse model of AD, the synaptic and cognitive defects characteristic of AD were improved by increasing the expression of Klotho [34] and this neuroprotective effect may depend on the ability of Klothoto increase antioxidant enzymes [35].
Conclusion
Vitamin D is a major custodian of the phenotypic stability of the Ca2+ and redox signalling systems that are central players in many human diseases. Any reduction in Vitamin D levels will result in a decline in the phenotypic stability of these signalling systems resulting in elevated neuronal Ca2+ and ROS levels and this could contribute to the age-related decline in cognition and it may also act to trigger the onset of AD. It is proposed that this dysregulation of Ca2+ initiates the formation of the pathological Aβ oligomers to trigger the Aβ/ Ca2+ positive feedback loop (Figure 4) responsible for the onset of AD [2]. Such a scenario is entirely consistent with the fact the Vitamin D deficiency is such a strong risk factor for AD.
It is clear that the medical community should be more aware of the importance of Vitamin D because there is increasing evidence that maintaining normal levels of this critical hormone would markedly reduce the development of AD and many of the other major human diseases.
References
- Holick MF. Vitamin D: extraskeletal health. Rheum Dis Clin North Am. 2012; 38: 141-160.
- Berridge MJ. Vitamin D cell signalling in health and disease, Biochem. Biophys Res. Commun. 2015.
- Dampf-Stone A, Batie S, Sabir M, Jacobs ET, Lee JH, Whitfield GK, et al. Resveratrol Potentiates Vitamin D and Nuclear Receptor Signaling. J Cell Biochem. 2014.
- MacLaughlin J, Holick MF. Aging decreases the capacity of human skin to produce vitamin D3. J Clin Invest. 1985; 76: 1536-1538.
- Decuypere JP, Monaco G, Missiaen L, De Smedt H, Parys JB, Bultynck G. IP(3) Receptors, Mitochondria, and Ca Signaling: Implications for Aging. J Aging Res. 2011; 2011: 920178.
- Riascos D, de Leon D, Baker-Nigh A, Nicholas A, Yukhananov R, Bu J, et al. Age-related loss of calcium buffering and selective neuronal vulnerability in Alzheimer's disease. Acta Neuropathol. 2011; 122: 565-576.
- Annweiler C, Schott AM, Berrut G, Chauviré V, Le Gall D, Inzitari M. Vitamin D and ageing: neurological issues. Neuropsychobiology. 2010; 62: 139-150.
- Landfield PW. 'Increased calcium-current' hypothesis of brain aging. Neurobiol Aging. 1987; 8: 346-347.
- Foster TC. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell. 2007; 6: 319-325.
- Bodhinathan K, Kumar A, Foster TC. Redox sensitive calcium stores underlie enhanced after hyperpolarization of aged neurons: role for ryanodine receptor mediated calcium signaling. J Neurophysiol. 2010; 104: 2586-2593.
- Brewer LD, Thibault V, Chen KC, Langub MC, Landfield PW, Porter NM. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci. 2001; 21: 98-108.
- Brewer LD, Porter NM, Kerr DS, Landfield PW, Thibault O. Chronic 1alpha,25-(OH)2 vitamin D3 treatment reduces Ca2+ -mediated hippocampal biomarkers of aging. Cell Calcium. 2006; 40: 277-286.
- Latimer CS, Brewer LD, Searcy JL, Chen KC, Popovic J, Kraner SD, et al. Vitamin D prevents cognitive decline and enhances hippocampal synaptic function in aging rats. Proc Natl Acad Sci U S A. 2014; 111: E4359-4366.
- Koran ME, Hohman TJ, Thornton-Wells TA. Genetic interactions found between calcium channel genes modulate amyloid load measured by positron emission tomography. Hum. Genet. 2014; 133: 85-93.
- Przybelski RJ, Binkley NC. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function. Arch Biochem Biophys. 2007; 460: 202-205.
- Schlögl M, Holick MF. Vitamin D and neurocognitive function. Clin Interv Aging. 2014; 9: 559-568.
- Toffanello ED, Coin A, Perissinotto E, Zambon S, Sarti S, Veronese N, et al. Vitamin D deficiency predicts cognitive decline in older men and women: The Pro.V.A. Study. Neurology. 2014; 83: 2292-2298.
- Dubal DB, Yokoyama JS, Zhu L, Broestl L, Worden K, Wang D, et al. Life extension factor klotho enhances cognition. Cell Rep. 2014; 7: 1065-1076.
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008; 31: 454-463.
- Querfurth HW, Selkoe DJ. Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry. 1994; 33: 4550-4561.
- Itkin A, Dupres V, Dufrêne YF, Bechinger B, Ruysschaert JM, Raussens V, et al. Calcium ions promote formation of amyloid β-peptide (1-40) oligomers causally implicated in neuronal toxicity of Alzheimer's disease. PLoS One. 2011; 6: e18250.
- Pierrot N, Santos SF, Feyt C, Morel M, Brion JP, Octave JN. Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-beta accumulation. J Biol Chem. 2006; 281: 39907–39914.
- Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron. 2013; 79: 887-902.
- Bodhinathan K, Kumar A, Foster TC. Redox sensitive calcium stores underlie enhanced after hyperpolarization of aged neurons: role for ryanodine receptor mediated calcium signaling. J Neurophysiol. 2010; 104: 2586-2593.
- Berridge MJ. Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion. 2013; 7: 2-13.
- Berridge MJ. Calcium regulation of neural rhythms, memory and Alzheimer's disease. J Physiol. 2014; 592: 281-293.
- Annweiler C, Fantino B, Le Gall D, Schott AM, Berrut G, Beauchet O. Severe vitamin D deficiency is associated with advanced-stage dementia in geriatric inpatients. J Am Geriatr Soc. 2011; 59: 169-171.
- Wang L, Hara K, Van Baaren JM, Price JC, Beecham GW, Gallins PJ, et al. Vitamin D receptor and Alzheimer's disease: a genetic and functional study. Neurobiol Aging. 2012; 33: 1844.
- Gezen-Ak D, Dursun E, Bilgiç B, Hanağasi H, Ertan T, Gürvit H, et al. Vitamin D receptor gene haplotype is associated with late-onset Alzheimer's disease. Tohoku J Exp Med. 2012; 228: 189-196.
- Sutherland MK, Somerville MJ, Yoong LK, Bergeron C, Haussler MR, McLachlan DR. Reduction of vitamin D hormone receptor mRNA levels in Alzheimer as compared to Huntington hippocampus: correlation with calbindin-28k mRNA levels. Brain Res Mol Brain Res. 1992; 13: 239-250.
- Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, et al. Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol. 2007; 66: 75-85.
- Kanninen K, Heikkinen R, Malm T, Rolova T, Kuhmonen S, Leinonen H, et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009; 106: 16505-16510.
- Ghosh D, LeVault KR, Brewer GJ. Dual-energy precursor and nuclear erythroid-related factor 2 activator treatment additively improve redox glutathione levels and neuron survival in aging and Alzheimer mouse neurons upstream of reactive oxygen species. Neurobiology of Aging. 2014; 35: 179-190.
- Dubal DB, Zhu L, Sanchez PE, Worden K, Broestl L, Johnson E, et al. Life extension factor klotho prevents mortality and enhances cognition in hAPP transgenic mice. J Neurosci. 2015; 35: 2358-2371.
- Zeldich E, Chen CD, Colvin TA, Bove-Fenderson EA, Liang J, Tucker Zhou TB, et al. The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J Biol Chem. 2014; 289: 24700-24715.