
Review Article
Austin J Clin Neurol 2015;2(5): 1045.
From Dark to Bright to Gray Sides of Memory: In Search of its Molecular Basis & Alzheimer’s Disease
Carlos Velez-Pardo* and Marlene Jimenez-Del-Rio*
Neuroscience Research Group, Medical Research Institute, University of Antioquia (UdeA), Colombia
*Corresponding author: Velez-Pardo C, Neuroscience Research Group, Medical Research Institute, School of Medicine, University of Antioquia (UdeA), Calle 70 No. 52-21, and Calle 62 # 52-59, Building 1, Room 412; SIU Medellin, Colombia
*Corresponding author: Jimenez-Del-Rio M, Neuroscience Research Group,Medical Research Institute, School of Medicine,University of Antioquia (UdeA), Calle 70 No. 52-21, and Calle 62 # 52-59, Building 1, Room 412; SIU Medellin,Colombia
Received: March 26, 2015; Accepted: May 19, 2015; Published: May 25, 2015
Abstract
Memory is one of the most fascinating functions of the brain. Without it, the human being condition would be lost. Therefore, alterations of memory are at the center of research. In this review, the most prominent memory case disorders are examined to identify basic differences and commonalities of the memory processes altered in the human brain. Then, relevant aspects of the molecular mechanism of memory between Aplysia, Drosophila, and mammals (mice) are highlighted in order to understand the biological aspect of memory in humans. The convergence of both topics provides a foundation for an integrative study of prevention and loss of memory in familial Alzheimer’s disease (FAD). Therefore, we propose that highly superior autobiographical memory (HSAM) and familial Alzheimer’s disease (FAD) are opposite extreme cases of “normal” memory and that their pathophysiology can be explained by changes in protein expression of the PKA / CREB-1 / CPEB axis. We also propose that Aβ directly intermingles with the CPEB. As a result of “yin-yang” prion-like protein interactions, Aβ is capable of interfering with the CPEB’s normal function. If validated, this hypothesis may help explain why anti-amyloid therapies have been negative or inconclusive so far. Therefore, therapies targeting intracellular Aβ oligomers are urgently needed. Molecular studies on HSAM individuals might be invaluable to discover molecules to increase memory skills in FAD.
Keywords: Alzheimer’s disease; CREB-1; CPEB; Explicit memory; Memory; HSAM; Implicit memory; Short-term memory
Abbreviations
AC: Adenylate Cyclase; AMPAR: α-amino-3-hydroxy-5-methyl- 4-isoxazolepropionic Acid receptor; C/EBP: CCAAT-box-enhanced Binding Protein; CaMKII: Ca2+/Calmodulin Protein Kinase II; CaMAC: Ca2+/calmodulin-activated Adenylyl Cyclase; Ca-i-PKC: Ca2+-independent PKC; CN: Calcineurin; CPEB: Cytoplasmic Polyadenylation Element Binding Protein; DAR: Dopamine Receptor; PLC: Phospholipase C; PP1: Protein Phosphatase-1; PKA: Protein Kinase A; PKC: Protein Kinase C; NMDAR: N-methyl-Daspartate Receptor; UH: Ubiquitin Hydroxylase; 5-HT-R: serotonin receptor
Introduction
Memory is the ability of the brain to encode, store, retain, and recall information including facts, experiences, impressions, skills, and habits. It gives living things the capability to learn (i.e., the process of acquiring knowledge of the world and adapting from previous experiences to affect or influence current behavior). Etymologically, the word “memory” derives from the Latin word memory and memoir, or from the Greek word thymesis meaning “mindful” or “remembering.”Since memory is an important part of our most intimate self-realization, philosophers, medical doctors, and lately, scientists have tried to understand what memory is, how it works, and why it goes wrong. Aristotle (384 a. C.-322 a. C.) was the first to compare memory to making impressions in wax, and suggested the idea that memories are copies of reality that a person stores and later retrieves, sometimes referred to as the “storehouse metaphor,” –a theory of memory that influenced thinking for many centuries.
However, it was not until the mid-1880s that the young German psychologist Herman Ebbinghaus (1850-1909) developed the first scientific approach to studying memory. Using himself as the research subject, Ebbinghaus was able to establish the shape of the learning and forgetting curve where he discovered that early and late items in a list are more likely to be recalled than middle items (i.e., primacy and recency effects), and reported that even a small amount of initial practice, far below that required for retention, can potentially avoid the need of re-learning. Ebbinghaus also classified memory into three distinct types: sensory memory (SM, the ability to retain impressions of sensory information received through the five senses); short-term memory (STM, the capacity to hold a small amount of information in the mind in an active, readily available state for a short period of time, in the order of 20-30 seconds up to 1 min); and long-term memory (LTM, the capacity to hold an indefinite amount of information for a longer period of time, in the order of days, weeks, or years) [1]. This classification remains relevant to this day.
Furthermore, studies by George A. Miller (1920-2012) in the mid-50s demonstrated that STM is limited to what he called “the magical number seven, plus or minus two,” reflecting an STM capacity of 7 ± 2 elements [2]. By 1972, the experimental psychologist Endel Tulving (1927-present) was the first to propose two distinct types of LTM: (i) episodic, defined as the collection of past personal experiences– autobiographical events, such as time, places, associated emotions, and other contextual particulars that can be explicitly stated; and (ii) semantic, which refers to the memory of meanings, understandings, and other concept-based knowledge [3]. Both semantic and episodic memory constitutes the category of declarative memory. Declarative memory– sometimes referred to as explicit memory– refers to memories that can be consciously recalled; whereas non-declarative or procedural memory- also referred to as implicit memory- is the second category of memory in which previous experiences aid the performance of a task without conscious awareness of these previous experiences [4,5].
The most popular model for studying memory- the multi-store or modal model of memory proposed by Atkinson-Shiffrin in 1968 [6] is depicted in Figure 1A-B. This model proposes memory as a sequence of three stages, from SM to STM to LTM (blue filled arrows). The brain regions (marked in red bold) used to stack memory processes such as encoding (process by which the perceived item of interest is converted into a construct that can be stored, and then recalled later from STM to LTM), consolidation (process of stabilizing a memory trace after initial acquisition), storage (process of retaining information in either SM, STM, or LTM, but mostly in LTM), and retrieval (process of reaccessing events or information from the past previously encoded and stored) are shown in purple bold. Although the understanding of human memory has benefited from studies on animal models of dementia and cognitive dysfunction [7], several studies indicate that memory is a highly complex function of the brain for which the exact (neural and molecular) mechanisms of action remain elusive. Therefore, except for the possibility of significantly reducing aversive memory by pharmacological treatment [8], therapies or over-thecounter medicines which maintain, improve, or avoid/stop loss of memory are not currently available.
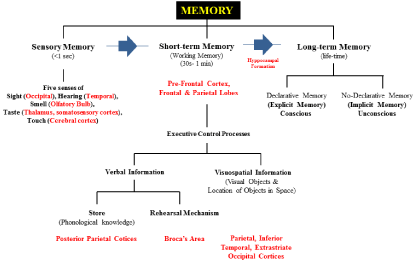
Figure 1A: A schematic representation of the types of memory and region
localization in the human brain.
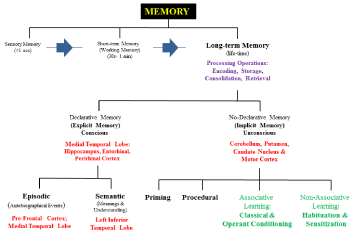
Figure 1B: A schematic representation of the types of memory and region
localization in the human brain.
We will examine the most prominent memory disorders, (e.g., cases of ultra-superior memory, savant syndrome, clinical cases of severe amnesia and dementia (e.g., Alzheimer’s disease), to determine basic differences and commonalities of the memory processes altered in the human brain.
The dark side of memory
“What have you done to my memory?” H.M.
On September 1, 1953, the neurosurgeon William B. Scoville performed a bilateral resection of the medial temporal lobes (MTL) on Henry Molaison (1926 – 2008), known as patient H.M. in scientific literature, as an experimental approach to alleviate a pharmacological intractable epilepsy [9]. The resection extended posteriorly for a distance of 54.5 mm and 44.0 mm in the left and right hemispheres, respectively [10]. Although the surgery reduced the frequency of his pre-operatively severe epileptic attacks and enabled their control with drugs(e.g., phenytoin or Delantin®), H.M. thereafter showed severe antero grade amnesia, (i.e., he no longer was able to form new memories despite the fact that his short-term memory (or attention span) was intact [9], and partial retrograde amnesia, (i.e., he lost his memory access to events that occurred or information that was learned up to three years before surgery). Recently, the exact anatomical areas removed by Scoville have been reported to include the medial temporopolar cortex (Brodmann Area #38), Piriform cortex, entorhinal cortex (BA#28), perirhinal cortex (BA#35) and subiculum (BA#27), the amygdala, anterior half of the hippocampus (BA#28), and the dentate gyrus. The posterior parahippocampal gyrus (BA#20) and medial temporal stem were partially damaged [11].
By 1962, Milner reported the first experimental demonstration of the dissociation between the acquisition of declarative memory and other kinds of learning (e.g., motor learning). Indeed, H.M. learned to draw the outlines of a star while looking at the star and his hand in a mirror. Clearly, H.M.’s time and error scores decreased within and across three days of training on a mirror-tracing task [12]. Later work has shown that H.M. preserved others kinds of learning abilities [13,14]. Furthermore, H.M. displayed adequate social skills (excessively polite), preserved normal verbal skills, and exhibited other normal psychological features (Table 1).
![]()
Subject
Brain Findings
Characteristics of the Persona&
Psychological Findings
The dark side of memory
Henry Molaison,
« H.M. » patient
1953: “Bilateral medial temporal lobe resection was carried out, extending posteriorly for a distance of 8 cm from the midpoints of the tips of the temporal lobes, with the temporal horns constituting the lateral edges of resection…” (Scoville and Milner, 1957).
1997: The MRI studies indicated that the bilateral medial temporal lobe lesion included “the medial temporal polar cortex, most of the amygdaloid complex, most or the entire entorhinal cortex, and approximately half of the rostrocaudal extent of the intraventricular portion of the hippocampal formation (dentate gyrus, hippocampus, and subicular complex)”. The length of the lesion was 54.0 mm and 51.0 mm in the left and right hemispheres, respectively. Half of the hippocampus body (caudal 20 mm) was intact, but atrophic (Corkin et al., 1997).
2013: 3D anatomical measurements in the medial temporal lobe showed that the length of the lesion was 54.5 mm and 44.0 mm in the left and right hemispheres, respectively. The extent of the spared hippocampus was 23.6 and 24.3 mm in the left and right hemispheres, respectively. At autopsy, multiple white matter lesions consistent with lacunar infarctions were present and Cerebellar atrophy was evident (Annese et al., 2014). Based on MRI data, the anatomical areas removed to H.M. patient were the medial tempopolar, piriform, and entorhinal cortices, subiculum, the amygdala, anterior half of the hippocampus, and the dentate gyrus (Augustinack et al., 2014).
“[H.M.] appears to have a complete loss of memory for events subsequent to bilateral medial temporal lobe resection 19 months before, together with a partial retrograde amnesia for the three years leading up to his operation”; displayed normal intelligence (IQ above 100) with no major emotional or behavioral changes; had a sense of self by stating, for example, What have you done to my memory? (Scoville and Milner, 1957). Time appeared to have stopped to him; he was unaware of the events happening in the world and in his own life; actually, “[H.M.] could no longer remember the faces of people he met, places he visited, or moments he lived through. His experiences slipped out of his consciousness seconds after they happened.” (Corkin, 2013). However, he was able to recognize faces of persons who had become famous in different decades (1920-1970) before his operation. Therefore, the MTL is not the ultimate storage site for previously acquired knowledge.
Clive Wearing (C.W.)
1991: The MRI and scan studies indicated that viral encephalitis destroyed extensive areas of the left-brain and the right mesial temporal lobe including hippocampal formation (both sides), and pre-frontal cortex. The Corpus callosum’s volume was importantly reduced and remnant of Fornix was left over.
C.W. suffers from severe anterograde and retrograde amnesia. He is confined to life in an eternal present time with neither past memories nor future thoughts .He suffers delusion, i.e., C.W. permanently believes he has just woken up for the first time, he seems to awake at two-or three-minute intervals by writing, for example, “Today: 1st CONSCIOUSNESS...Conscious for the FIRST TIME” or“ I am
completelyawake” over and over again with minor modifications; he has no knowledge of where he was or what had happened to him“ I have nothing to say about it,..., no dreams, no thoughts, nothing at all… It’s like being dead …”; he has retained a tacit understanding of social conventions, he has apparently no sense of self, and no sense of reality.Augusta Deter,
« A.D. » patient
1906:“The autopsy revealed an evenly atrophic brain without macroscopic foci. The large cerebral vessels show arteriosclerotic changes…In preparations stained with Bielschowsky’s silver method, peculiar changes of the neurofibrils manifest themselves…. Then these fibrils aggregate together into dense bundles and emerge eventually to the surface of the cell...Scattered over the entire cortex, especially numerous in the upper layers, miliary foci can be found, which are caused by deposition of a peculiar substance in the cerebral cortex. It is recognizable without staining and is rather refractory to staining…” (Alzheimer, 1906).
“A woman of 51 years showed jealously of her husband as the first obvious symptoms of the disease. Soon fast increasing memory impairment became apparent; she lost orientation in her own home; carried objects around and hid them; sometimes she believed someone was going to murder her and she started to scream loudly…She is completely disoriented in time and space. Occasionally she remarks that she does not understand anything and nothing is familiar to her...At times she is totally delirious,…, and she seems to have auditory hallucinations…Her capacity of memory is most severely impaired. If one shows objects to her, she is usually able to name them correctly, but immediately after forgets everything…” (Alzheimer, 1906).
Table 1: The most relevant cases of memory disorders, along with brain findings, characteristics of the persona and psychological findings.
The significance of the reported case of H.M.’s amnesia was fivefold. First, it established the fundamental principle that memory is a distinct cerebral function. Second, it provided compelling evidence that the declarative (explicit) memory system is critically dependent upon the medial temporal region, and that non-declarative (implicit) memory involved in learning was located in other brain regions. Furthermore, it clearly demonstrated that the hippocampal formation is necessary and sufficient to form long-term memories. Third, it suggested a positive relationship between the extent of destruction of the hippocampal formation and the degree of memory impairment, specifically loss of recent memory. Fourth, H.M. was able to retain information for a time after it was presented (e.g., he could retain a three-digit number as long as 15 min by continuing rehearsal) and was able to carry on a conversation, which supports an important distinction between short-term memory and long-term memory. Finally, it motivated not only the development of animal models of human amnesia [15], but also inspired the study of the molecular bases of memory (see, for example, the 2000 Nobel prize of Eric Kandel for “discoveries concerning signal transduction in the nervous system”) [16] and placement memory (see the recent Nobel Laureates John O’Keefe, May-Britt Moser, Edvard I. Moser, winners of the Nobel Prize in Physiology and Medicine 2014 for “their discoveries of cells that constitute a positioning system in the brain” [17-19]). Therefore, the description of H.M. is perhaps the most relevant case in the history of neuropsychological research [20,21], and marks the unprecedented beginning of scientific research aimed at understanding how the human brain works.
“I am incapable of thinking” C.W.
On March 27, 1985, Clive Wearing (1938 - present) contracted herpes viral encephalitis, a herpes simplex virus that attacked his central nervous system. As a result, Wearing suffers from severe anterograde and retrograde amnesia. This has become a remarkable case of dense amnesia [22,23]. Wearing’s case was made public in the 1986 documentary entitled Equinox: Prisoner of Consciousness (director John Dollar). Presently, he is tenderly known as “The man who keeps falling in love with his wife” [24], or even more dramatically as “The Man with a 7 second memory.” According to Dr. E. Bigler (Brighman Young University), who analyzed the MRI and brain scans in 1991, viral encephalitis destroyed a substantial portion of his left-brain and mesial temporal lobe on his right brain side. There was clearly enlargement of the ventricular system as an indication of the generalized damage of the brain. Strikingly, encephalitis bilaterally devastated the hippocampal formation and the pre-frontal cortex. Because the lesions were asymmetrical, it is difficult to know exactly to what extent the left extra hippocampal damage contributes to his neuropsychological findings (Table 1).
Despite the fact that Wearing’s amnesia includes semantic as well as episodic memory deficits, he is still able to play the piano, read musical notes and sing; C.W. is a British musicologist, conductor, tenor, and keyboardist. As noted by Jiri Rezac (photographer), “Clive at the keyboard, his musical powers remain intact.” This last observation suggests that the patient still preserves his implicit (procedural) memory. In 2007, the neurologist Dr. Sacks visited Clive Wearing and dramatically concluded that “It has been twenty years since Clive’s illness, and for him, nothing has moved on. One might say he is still in 1985 or, given his retrograde amnesia, in 1965. In some ways, he is not anywhere at all; he has dropped out of space and time altogether. He no longer has any inner narrative; he is not leading a life in the sense that the rest of us do” [25].
Clearly, Clive Wearing’s case goes beyond clinical interest. What are the implications for those that study the body-mind problem? Specifically, what is the relationship between consciousness, “soul”, and the brain? Is Clive’s case a serious consideration for denying the religious idea of God? Or, does it simply support the notion that life is purely physical biochemistry governing the brain and its mind (i.e., a global brain function), rather than spiritual? Future studies are needed to elucidate these issues. Global amnesia sufferers vividly show us that bilateral MTL damage separates the human being from properly interacting with the environment.
“I have lost myself” A.D.
Augusta Deter (1850 – 1906) is, without a doubt, the most cited patient associated with Dr. Alois Alzheimer, a German psychiatrist who in 1901 diagnosed her as suffering from “disease of forgetfulness.” By the time Dr. Alzheimer examined her, the patient not only displayed cognitive impairment and aphasia, but also typical psychiatric symptoms such as delusions, auditory hallucinations, sleep disorder, paranoia, unpredictable behavior, and focal symptoms (i.e., reduced comprehension, disorientation, “amnestic writing disorders” and loss of memory) [26] for a detailed description of the file of A.D., see ref [27].
Despite the fact that she would have been diagnosed with a psychiatric disorder, Alzheimer pointed out three odd features in A.D. First, she had no sense of time or place. Second, she was relatively young (51 years) to present substantial loss of memory and senile dementia. Finally, at her death (56 years), she presented atypical brain histopathology– neurofibrillary tangles and “deposition of a peculiar substance in the cerebral cortex,” (i.e., the amyloid-β (Aβ)-plaques identified by Glenner and Wong in 1986) [28] (Table 1). A.D.’s disorder and others similar to hers is now well known as “Alzheimer’s disease (AD).”
Interestingly, more than 100 years after this case was reported [26], a DNA genetic analysis of a histological section of A.D. demonstrated that a mutation in the presenilin-1 (PSEN-1) gene (c. 526T>C, p. Phe176Leu, exon 6) was the cause of the disease [29]. AD is now recognized to be familial (<5% of cases by mutation in at least three genes: amyloid β precursor protein (AβPP), PSEN-1 and PSEN-2; https://www.molgen.vib-ua.be/ADmutations/) and sporadic in origin. Currently, AD is defined as a progressive, neurodegenerative disorder that progresses through three disease stages over the course of 5 to 20 years: asymptomatic (pre-clinical or silent stage of AD), mild cognitive impairment (MCI), dementia (Alzheimer’s proper) [30]. MCI is a transitional state between “normal” forgetfulness and dementia, and when memory is affected, the condition is called amnesic MCI. Indeed, while anterograde amnesia (i.e., loss of episodic memory) is the clinical hallmark in MCI patients, both anterograde and retrograde amnesia, as well as the loss of working memory is characteristic of AD. Accompanying cognitive and behavior progressive changes [31] are profound alterations in brain morphology [32] and histology [33] (Figure 2A vs. 2B). Progressive neurodegeneration includes the accumulation of Aβ peptides, mainly 42 amino acids long (Aβ1-42) in extracellular senile plaques, and occasionally in blood vessels, and the buildup of hyperphosphorylated forms of the microtubule-associated protein Tau in intracellular neurofibrillary tangles.
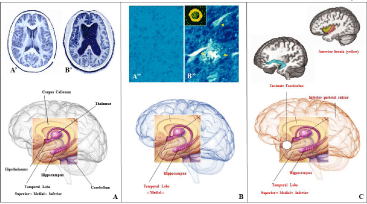
Figure 2: A schematic representation of normal (A), Alzheimer’s (B), and high superior autobiographical memory (C) brain along with unaffected (bold black) or
affected (bold red) regions. Magnetic resonance imaging (MRI) of normal (A’) and Alzheimer’s disease (B’) brain. Histopathological findings in normal (A’’) and
Alzheimer’s disease (B’’) brain tissue stained with Thioflavine-S (Arrow, senile plaque; arrowheads, neurofibrillary tangles).
Elucidating the cognitive and biochemical changes that accompany these changes remains an area of intense investigation [34-36]. Because failure of clinical trials has been attributed to testing of potential disease-modifying therapeutic agents too late, research has focused on identifying the most prominent and earliest cognitive, behavioral, and structural brain symptoms to early intervention. By using voxel-based morphometry (VBM) and shape analysis of magnetic resonance imaging (MRI) data, it has been shown that individuals with pre-clinical AD and MCI have significantly reduced brain volume, not only in the medial temporal lobes, but also in the posterior cingulate/precuneus, and orbitofrontal cortex compared with control individuals [37]. Moreover, using VBM cross-sectional analysis, it has demonstrated that in progressive MCI, patients revealed cerebral areas of lower grey matter volume in the parahippocampal gyrus, precuneus, and posterior cingulate 12 months prior to AD diagnosis [38]. Recently, Khan et al. [39] have implicated the lateral entorhinal cortex as the earliest area of the brain to be affected in pre-clinical AD.
Taken together, these observations comply with the notion that structural brain changes occur (10-12) years before clinical, cognitive decline in AD [40,41], and that brain changes are indicative of decreased synaptic density, neuronal loss, brain atrophy, which interrupt the neuronal network, which is critical for declarative (episodic) memory [42]. However, others have found greater atrophy in the hippocampus, predominantly in the CA1 region and subiculum and entorhinal cortex [43-45].
These discrepancies in diagnostic accuracy might reflect either differences between the methods used [46] or variability of which marker is used (i.e., amyloid imaging, 18F-fluorodeoxyglucose [FDG]-PET, SPECT, MRI) as well as how it is measured (“metric:” visual, manual, semi-automated, or automated segmentation/ computation) [47]. Furthermore, a number of challenges must be overcome before the medical community can attempt to intervene identified pre-clinical subjects to delay and even prevent emergence of the clinical syndrome [48]. Therefore, further investigation is needed to clarify these issues.
Recognizing that, the pathophysiological process of AD begins years, if not decades, prior to clinically obvious symptoms (i.e., preclinical stage of AD) calls into question the definition of AD. Simply put, the difference lies in whether AD is really a neurodegenerative disease or whether it better qualifies as a neurodevelopmental disorder, especially in those subjects who carry fully penetrant genetic alterations that result in familial Alzheimer’s disease (FAD) (e.g., Colombian kindred bearing with autosomal dominant/recessive mutation in PSEN-1 [49,50]). This last issue is a pressing one to be answered given its definitive impact on therapeutic outcomes and in memory research.
In conclusion, the study of the aforementioned cases (e.g., H.M., C.W., A.D) [22,26] and other relevant ones with amnesia (e.g., R.B., G.B., E.P) [51] generally reveal that inflicted damage to MTL from surgery, infection, neurodegenerative disease, or by other causes (e.g., stroke, tumor, drugs, hypoxia) disturbs the brain to form long- term memories. Amnesic patients, but not advanced AD patients, are still able to retain working (short-term) and implicit (procedural) memory regardless of MTL structures. Specifically, bilateral damage to any single component of the hippocampal formation, including the hippocampus (e.g., bilateral lesion of the entire CA1 field of the hippocampus), dentate gyrus, subiculum, and adjacent structures such as the entorhinal, perirhinal, and parahippocampal cortices, might be sufficient to produce clinically significant and readily detectable memory impairment.
Undoubtedly, the study of anterograde and retrograde amnesia has led to the traditional idea that MTL structures are involved in the formation of LTM and that immediate memory, which refers to the limited amount of information that can be held in the mind when material is presented for learning, and working memory are independent of these structures. However, Ranganath and Blumenfield [52] and Graham et al. [53] have revisited this idea. Basically, these authors proposed that MTL, in addition to its established role in forming LTM, is needed for at least some kinds of working memory, and that the perirhinal cortex is needed for certain kinds of visual perception. Jeneson and Squire (2012) and Jeneson et al. (2011) have in turn reappraised this notion [54]. They have reached the conclusion that performance on tasks depends on LTM even when the retention interval is brief.
Therefore, the authors have elegantly proposed that MTL lesions impair performance only when immediate memory and working memory (the capacity to maintain a limited amount of information through active rehearsal, usually across short time) are overloaded with information to support performance. Additionally, they introduce the notion that a short-term delay task (subspan memoranda) is supported by immediate memory and working memory and are independent of the MTL. Furthermore, the view that the hippocampus and other medial temporal lobe structures are linked to both memory and spatial cognition has also been reexamined. Kim et al. have shown that memory and spatial cognition can be two separate entities [55]. Indeed, the researchers carried out studies of path integration in patients with medial temporal lobe lesions. Individuals entered a circular arena without vision, searched for a target, and then attempted to return to the start location. The patients performed accurately as well as the controls. This result has been interpreted as their ability to construct a coherent working memory of spatial environments.
Additionally, it has been demonstrated that structures other than the hippocampus can support face recognition memory in patients with hippocampal lesions [56] and “fast mapping” tasks [57]. Interestingly, Smith and coworkers [58] have quantitatively found an orderly relationship between anterograde and retrograde amnesia, such that the patients bearing bilateral MTL lesions with more severe anterograde amnesia had more extensive retrograde amnesia. However, whether amnesia is due to a deficit of encoding, storage, or retrieval is not yet fully understood. Therefore, further investigation is needed to solve these issues. This is of paramount importance for therapeutic approaches to prevent, protect, and improve memory and cognitive performance not only in amnesic but also in AD patients.
The bright side of memory
“I run my entire life through my head every day and it drives me crazy!!!...” A.J.
Jill Price (1965 – present) is the first person to be recognized by the scientific (known as A.J.) and public (known as The woman who can’t forget, [59]) community to possess highly superior autobiographical memory (HSAM), a phenomenon previously known as hyperthymesic syndrome [60]. HSAM is a rare condition in which individuals are able to recall events from their personal past, including the days and dates on which they occurred, with very high accuracy. Indeed, Price can recall in vivid detail events (i.e., she can evoke what day of the week a particular date fell on within her mental calendar) from her past since age 12 in a “nonstop, uncontrollable, and automatic” fashion. Extensive neuropsychological studies have not only demonstrated Price’s memory superiority (e.g., General Memory Index on Wechsler Memory Scale-Revised: score = 122, 1.5 SD above average, Autobiographical Memory Index: score 27/27, (Table 1), but they have also revealed atypical brain lateralization (i.e., although Price insisted she is right-handed, she showed left-handed dominance on several tasks) and atypical variant in neuropsychological tests, which suggested some brain areas with noticeable strengths (e.g., medial temporal lobe) and weaknesses (e.g., pre-frontal and frontal lobe, anterior left hemisphere).
Despite these observations, she also displayed other neuropsychological domains (e.g., calculations, motor speed, visual, spatial, and language) within the normal range (within 1.5 SD). These findings were interpreted by Dr. McGaugh’s research team, who suggested that Jill Price might have a variant of a neurodevelopmental, frontostriatal system disorder which includes obsessive-compulsive disorder (OCD), thus explaining her diagnosis of hyperthymesic syndrome [60].
Further studies by the same research group have identified ten additional cases of HSAM individuals. The eleven individuals (including J.P.) excelled in several cognitive assessments, such as autobiographical memory task, recall of names paired with faces, visual memory test, logical memory free-recall test, and Leyton Obsessional Inventory Score-Short compared to control individuals. However, HSAM individuals showed no differences in forward and backward digit span test, visual reproduction test, and logical memory recognition test compared to control group [61].
Remarkably, the investigation revealed both behavioral and neuroanatomical domain commonalities among HSAM participants. First, it was verified that HSAM individuals have an impressive memory for autobiographical events and extensive knowledge of public event information. Second, for the typical HSAM participant, the range of dates for which the days and dates are recalled is limited to dates within their lifetime, and in particular from the age of 10.5 years old, a mean age at which HSAM individuals became aware of their ability to remember events from most days. Third, most of the HSAM participants (9/11) tend to display a degree of OCD. Fourth, as reported previously with the “A.J.” case, HSAM participants showed no differences in their performance on digit-span forward, verbalpaired associates, and visual reproduction, but performed significantly better than controls on the Logical Memory test free-recall and Names-to-Faces test. It was concluded that HSAM individuals “do not possess a domain-general, highly effective ability to encode and retrieve new information. Instead, it is more domain-specific as even those tests in the cognitive battery in which they outperform controls can be viewed in a personal, autobiographical manner.…” [61]. Finally, by using four structural imaging analyses (e.g., Voxel based morphometry-gray matter, voxel based morphometry-white matter, tensor based morphometry, diffusion tensor imaging-fractional anisotropy), unusual structural differences (i.e., expanded regions) were found in the HSAM participants in the region of the inferior and middle temporal gyri and temporal pole, the anterior insula, the parahippocampal gyrus, inferior parietal sulcus, uncinate fascicle, anterior putamen, caudate and posterior pallidum [61] (Figure 2C).
The authors suggested that it was highly plausible that all these regional brain differences, and especially the medial temporal lobe and uncinate fascicle, might not only contribute to explain the HSAM participants’ exceptional autobiographical memory, but also their behavioral trend towards OCD tendencies. In support of the former idea, it is noteworthy that patients with bilateral lesions to the medial temporal lobe (as discussed above for H.M. and C.W.) demonstrated retrograde memory loss, for both public and personal autobiographical facts and events.
Therefore, it is highly conceivable that HSAM individuals rely upon these regions to excel in autobiographical memory. Because HSAM individuals distinguish themselves by their ability to retain what they do learn, this provided an appealing framework in cognition-based intervention research (e.g., [62-64]. Molecularly, how the brain produces a “superior memory” is still a fascinating enigma.
“I have so many things to me that you can’t even guess them all” K.P.
Kim Peek (1951 – 2009) was born with macrocephaly, damage to the cerebellum, and agenesis of the corpus callosum. He was diagnosed with savant syndrome. Formally, the disorder is a rare but spectacular condition in which persons with developmental disabilities, including but not limited to autism, or other CNS disorders or diseases have some outstanding “island of genius” that stands in marked contrast to overall physical disability [65]. Notably, the extraordinary talents associated with prodigious savants are invariably linked to memory.
Particularly, K.P. is known as “mega-savant” because of his unlimited memory capacity. Given the fact that he read a page in 8- 10 seconds, it was suggested that Peek had learned at least 9,000 books recalling their content with 97% accuracy, decades later. Interestingly, he apparently understood the material he had memorized. K.P. also displayed numerous skills, including calendar calculating, lightningspeed calculation abilities, piano performance, and encyclopedic knowledge in several topics (e.g., world and American history, geography, literature, music, among others) [66]. Despite the fact that Kim’s memory itself could be considered his savant skill, he had trouble following directions and focusing, showed poor social skills and verbal communication skills, as described by the scientist he met in 2006 [67]. Moreover, one of his greatest difficulties was abstraction or conceptual thinking. Indeed, as concluded by Dr. V.S. Ramachandran (Neuroscientist, University of California, San Diego) and his research team after conducting several tests on K.P. to evaluate conceptual encoding: “…What’s exciting about these results is- and what it’s showing you already- is that Kim, it turns out, is not editing, censoring, and encoding the information in the same way that normal people do, and it’s this lack of conceptual encoding that makes some of these savants adhere slavishly to every little detail and actually improves their memory in some respects, but of course they are paying a price for it…”, furthermore, “…[his] tendency to take metaphors literally, is another example of a failure of conceptual encoding…”According to Dr. Rita Jeremy (University of California, San Francisco) “… Altogether, he is a very unusual profile that cannot be summed up in any single score…” [67].
Sadly, K.P. died of a heart attack in his hometown Salt Lake City at the age of 58. Kim inspired the movie “Rain Man”, a 1988 film directed by Barry Levinson, starring Dustin Hoffman and Tom Cruise, several (~19) documentaries, and a book written by his father captured his real nature [68]. However, in comparison to other important cases (e.g., [11,69]), not a single scientific article (to our knowledge) has been published in a peer-reviewed journal reporting K.P.’s case. However, it is revealing to hear Dr. V.S. Ramachandran ask him, “How do you do that?” [i.e., the memory process, indeed]. Kim fancifully addressed the answer to his father (Francis Peek) by asserting that “They are still unable to find out what all of this is about, aren’t they dad?” [67]. This is not surprising. Savants are puzzled too! And normally cannot explain how they perform their skills. To date, no information is available to establish whether Kim’s brain was preserved to perform further post-mortem histological analysis.
Still, several questions remain unanswered. What brain anatomical structure(s) provided Kim’s skills? Did the size, shape, and neuronal structure of his hippocampus deviate from normal? Was his neural network hyper-connected? Was Kim intelligent? Surely he was, but how then to define him? [70]. Unquestionably, K.P. was “single brained.” Is it possible to conciliate the assumption that savant abilities arise from left brain injury with right brain compensation in a unified brain, as found in Kim’s? Although recent experimental observations with transcranial direct current stimulation [71] and new “acquired” savant skills suggest that the theory is a valid lead for further investigation, it seems not to apply to Kim. Can humans attain the talent of a savant without sacrificing everyday function? Last, but not least, is it possible to design a “better” brain? [72,73]. To further complicate matters, a study conducted by Opitz and coworkers suggests that K.P. probably had FG syndrome [74]. FGS, also known as Opitz–Kaveggia syndrome [75] is a rare genetic syndrome caused by one or more recessive genes located on the X chromosome and causing physical anomalies and developmental delays including retardation, hyperactivity, hypotonia, a characteristic facial appearance and macrocephaly (e.g., [76]). However, neither cytogenetic analysis nor formal neuropsychological data nor additional diagnostic tests from Kim are available to confirm or dismiss such a claim.
We conclude that K.P.’s skills were probably due to the assembly of the four operations of explicit memory- “deep” encoding, storage, consolidation, and retrieval, in one processing unit; whereas J.P.’s HSAM might be caused by a highly-self focused storage and consolidation of episodic events, somehow “contaminated” or “intruded” by an overpowering unconscious retrieval of daily life futile details. We also conclude that in contrast to the majority of savant individuals, being HSAM does not modify professional development, social life, or lifestyle (e.g., Marilu Henner, actress; Aurelien Hayman, student at Durham University, UK; Louise Owen, violinist). Whether it would be a privilege or a burden to possess a superior memory is a personal viewpoint. According to Jill Price, this is an affliction.
Ultimately, these cases suggest that the way the brain is wired will probably determine how “successful” a person would be with his/her interpersonal relationships. We think that, despite the high complexity of our brain, which might be portrayed with the recurrent question “How do you do that?”in the cognitive field, Jill Price’s“ super memory” is a unique opportunity to enlighten basic (e.g., [77]) and pharmaceutical research (e.g., [78,79]) to define new methods to improve our memory focused on more practical and professional activities and, perhaps one day by the same means, to prevent memory loss in subjects with high risk of suffering from either familial or sporadic AD. Nonetheless, before this can happen, the molecular mechanisms of memory have to be fully elucidated and pharmaceutical targets have to be established. We review this last issue next.
Memory: in search of its molecular basis
As a pioneer of the biology of mind research, Dr. Kandel ER has used the living sea slug Aplysia californica to demonstrate the molecular, cellular, and circuit mechanisms that underlie the learning and memory processes. During the last 15 years, he has unprecedentedly popularized the molecular basis of memory, (i.e., How memories are made, stored, retrieved, and lost in the renowned textbook chapter “Cellular mechanisms of implicit memory storage and the biological basis of individuality”, published in the Principles of Neural Science [80]); and this topic has been reviewed extensively [81-88]. Also, an inspiring historical account of a personal quest to unlock the biological basis of memory is outlined in his book “In search of memory. The emergence of a new science of mind.” [89].
Several outstanding observations from Kandel’s work can be highlighted. First, based on stimuli of the siphon and/or tail and gill-withdrawal reflex response in Aplysia, he and his team worked to establish the cellular and molecular mechanisms of habituation, sensitization, and classical conditioning. Basically, habituation is the result of drastic decrease in the strength of synaptic transmission from excitatory (glutamatergic) sensory neurons, interneurons, or both to the motor neurons; depending on the duration of stimuli, it can lead to short-term or long-term habituation. Interestingly, anatomical studies showed that long-term habituation resulted from a decrease in the number of synaptic contacts between sensory and motor neurons [90]. In contrast, sensitization results from an increase in synaptic transmission at several connections in the neural circuit made by sensory neurons with serotonergic interneurons and motor neurons. Through serotonin binding receptor (SBR, neurotransmitter receptor (NTR) in Figure 3) coupled to G proteins, serotonin (5- HT; neurotransmitter (NT) in Figure 3) triggers the activation of two parallel pathways: 5-HT > SBR> Gs > adenyl cyclase (+ATP) > cAMP > cAMP-dependent PKA > K+ channels > Ca2+; and 5-HT > SBR> Gq/11 >PLC > diacylglycerol > PKC. As noted, activation of PKA and PKC kinases results in sustained release of neurotransmitters (glutamate) from the sensory neuron to motor neuron, and a strong gill-withdrawal reflex is induced. Sensitization can also be short-term or long-term. In classical conditioning, a withdrawal reflex of Aplysia is greater and longer-lasting enhancement. Classical conditioning involves two paired stimuli: a conditioned stimulus (CS), which is harmless to the organism (e.g., a light, a tone, a touch), and produces no overt response unrelated to the response that will be learned; an unconditioned stimulus (US), which is also harmless, but induces a strong and sustained response (e.g., salivation). Importantly, repeated pairing of the CS and US causes the CS to become an anticipatory signal for the US.
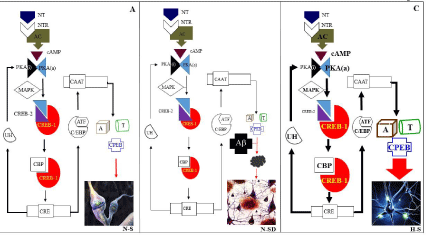
Figure 3: A schematic representation of the molecular mechanism of memory consolidation in normal (A), Alzheimer’s disease (B), and high superior autobiographical
memory(C). For detail, see text.
Abbreviations: NT: Neurotransmitter; NTR: Neurotransmitter Receptor; AC: Adenylyl Cyclase; PKA(a) or PKA(i): Protein Kinase A Active or Inactive; MAPK:
Mitogen-activated Protein Kinase; CREB-1 and CREB-2: cAMP Response Element Binding Protein; CBP: CREB-binding Protein; CRE: cAMP Response Element;
CAAT: CAAT Box; C/EBP: CAAT Box Enhancer Binding Protein; ATF: Activating Transcription Factor; A: Actin; T: Tubulin; CPEB: Cytoplasmic Polyadenylation
Element-binding Protein; Aβ: Amyloid-beta; ND-SD: Neuronal Death-synaptic Dysfunction; NS: Normal-synapsis; HS: Hyper-synapsis
Second, Kandel’s lab demonstrated that the conversion of short- term memory into long-term memory (i.e., consolidation) requires de novo messenger RNAs, and protein synthesis in the neurons (sensory neurons) and in facilitating interneurons (sensory neuron- motor neuron circuit), producing the growth of new synaptic connections between sensory neurons and motor neurons. Briefly, the mechanism involved in the prolonged activation of PKA via 5-HT/ SBP/Gs, allows the catalytic subunit of PKA kinase to translocate into the nucleus of the sensory neurons. Once there, active PKA in the presence of ATP activates mitogen-activated protein kinase (MAKP), which phosphorylates the transcription repressor CREB-2 (cAMP response element binding protein) liberating the transcription factor CREB-1. This unrepressed factor, in turn, binds a promoter element, CRE (cAMP response element), which together with the coactivator CREB-binding protein (CBP) turn on specific genes. Two important genes are essentially expressed for long term-facilitation: ubiquitin carboxiterminal hydrolase, which facilitates ubiquitinmediated protein degradation, thereby increasing activation of PKA; and the transcription factor C/EBP (CAAT box enhancer binding protein), which can form both homodimers with itself and form heterodimers with activating transcription factor (e.g., ATF4). These complex factors, in turn, act on downstream genes such as N-actin and tubulin, which cause the growth of new synaptic connections that support long-term memory (Figure 3A).
Third, it was found that alterations in the molecular component(s) of long-term memory not only affect Aplysia but also other model systems such as mice/rat and fly Drosophila melanogaster. Indeed, it has been found that either up-regulation of dominant active CREB- 1 mutants in mice [91], or expression of constitutively active CREB protein in Aplysia [92] and in rat [93,94] increase long-term synaptic potentiation/plasticity and memory storage. In contrast, inhibition of CREB-1 by CREB-2 in Drosophila [95], inhibition of ATF and C/EBP [96] or expression of a truncated form of CBP in mice [97] exhibited deficit in synaptic plasticity and memory storage.
Together, these observations suggest that the machinery for shortterm and long-term memory is conserved through evolution (Table 2). Fourth, Kandel and co-workers elegantly explained the molecular mechanism implicated in storage for long-term memory. They found that 5-HT increases (by an undetermined mechanism involving PKA and Phosphoinositide 3-kinase, PI3K) the synthesis of Aplysia CPEB (cytoplasmic polyadenylation element-binding protein) at the synaptic terminal of sensory neurons, working as a regulator of local protein synthesis [98]. Interestingly, the induction of CPEB is independent of transcription, probably via PI3K, but requires new protein synthesis via PKA. Most intriguingly, it was found that CPEB has prion-like properties, (i.e., self-perpetuating molecule [99,100]). Recently, Raveendra et al. have shown that Aplysia CPEB can exist as an alpha-helix–rich and a beta-sheet–rich amyloid (aggregated) conformational structures [101].
![]()
Specie/
Protein
Name
Homo
sapiens
Accession
number
Aplysia
californica
Accession
number
Drosophila
melanogaster
Accession
number
Hs vs Ac
BLAST result:
Identity, %;
Similarity, %; (E)
Hs vs Dm
BLAST result:
Identity, %;
Similarity, %; (E)
CaMAC
AC type 1
Q08828.2AC
NP_001191588.1
Ca(2+)/calmodulin
-responsive
adenylate cyclase
P32870.2
40%; 57%; (0.0)
46%; 64%; (1x10-112)
ACtype 8
P40145.1AC
NP_001191588.1
Ca(2+)/calmodulin
-responsive
adenylatecyclase
P32870.2
40%; 58%; (0.0)
58%; 76%; (6x10-139)
PKA
PKA alpha
(beta)
P17612.2
Catalytic subunit of PKA
NP_001191420.1
PKA c1
Isoform B (C,D)
AAN10703.1
85%; 92%; (0.0)
82%; 89%; (0.0)
PKA gamma
P22612.3
P22612.3
P22612.3
Catalytic subunit of PKA
NP_001191420.1
PKA c1
Isoform B (C,D)
AAN10703.1
74%; 87%; (0.0)
75%; 85%; (0.0)
PKC
PKCepsilon
CAA46388.1
Ca-i-PKC
NP_00119140.1.1
PKC
P13678.1
63%; 77%; (0.0)
60%; 74%; (0.0)
PKC alpha
EAW89014.1
Ca-i-PKC
NP_00119140.1.1
PKC
P13678.1
64%; 78%; (1x10-155)
63%; 79%; (8x10-154)
PKC gamma
EAW72161.1
Ca-i-PKC
NP_00119140.1.1
PKC
P13678.1
58%; 75%; (3x10-129)
44%; 57%; (8x10-144)
PKC eta
AAH37268.1
Ca-i-PKC
NP_00119140.1.1
PKC
P13678.1
54%; 69%; (0.0)
55%; 68%; (0.0)
PKC beta
AAH36472.1
Ca-i-PKC
NP_00119140.1.1
PKC
P13678.1
61%; 76%; (1x10-149)
63%; 70%; (1x10-153)
PKC iota
AAB17011.1
Ca-i-PKC
NP_00119140.1.1
PKC
P13678.1
43%; 58%; (1x10-135)
43%; 57%; (3x10-136)
PKC zeta
AAA36488.1
Ca-i-PKC
NP_00119140.1.1
PKC
P13678.1
44%; 58%; (3x10-129)
43%; 60%; (6x10-135)
PKCtheta
AAI13360.1
Ca-i-PKC
NP_00119140.1.1
PKC
P13678.1
43%; 58%; (0.0)
46%; 62%; (0.0)
PKC mu
CAA53384.1
Ca-i-PKC
NP_00119140.1.1
PKC
P13678.1
30%; 49%; (8x10-31)
30%; 49%; (6x10-31)
PP-1
PP-1
AAA36508.1
PP-1
XP_005105569.1
PP-1
CAA39820.
42%;54%; (0.018)
88%; 92%; (0.0)
UH
UHisozyme L1
P09936.2
UH
NP_001191493.1
UH36
Q9VRP5.3
43%; 62%; (1x10-58)
50%; 70%; (8.9)
PLC
PLC beta1
AAF86613.1
PLC
NP_001191617.1
PLC
AAA28820.1
38%; 54%; (4x10-45)
50%; 68%; (1x10-140)
PLC beta2
AAP35551.1
PLC
NP_001191617.1
PLC
AAA28820.1
33%; 53% (8.6)
41%; 60%; (2x10-48)
PLC beta3
CAA85776.1
PLC
NP_001191617.1
PLC
AAA28820.1
38%;55%; (2x10-44)
46%; 63%; (0.0)
PLC beta4
AAI43869.1
PLC
NP_001191617.1
PLC
AAA28820.1
37%; 56% (2x10-49)
46%; 64%; (3x10-128)
PLC delta1
AAA73567.1
PLC
NP_001191617.1
PLC
AAA28820.1
38%;55%; (2x10-73)
46%; 59%; (1x10-58)
PLC delta3
AAH72384.1
PLC
NP_001191617.1
PLC
AAA28820.1
36%;51%; (2x10-62)
35%; 52%; (2x10-55)
PLC delta4
AAH06355.1
PLC
NP_001191617.1
PLC
AAA28820.1
37%;56%; (2x10-75)
45%; 61%; (4x10-63)
PLC epsilon1
AAI51855.1
PLC
NP_001191617.1
PLC
AAA28820.1
34%; 52%; (1x10-38)
42%; 58%; (6x10-43)
PLC gamma1
AAI44137.1
PLC
NP_001191617.1
PLC
AAA28820.1
32%; 50%; (2x10-45)
44%; 58%; (6x10-47)
PLC gamma2
AAQ76815.1
PLC
NP_001191617.1
PLC
AAA28820.1
31%; 50%; (4x10-42)
34%; 50%; (2x10-49)
PLC eta
AAI13951.1
PLC
NP_001191617.1
PLC
AAA28820.1
41%;60%; (2x10-83)
29%; 46%; (5x10-88)
PLC zeta
AAN71895.1
PLC
NP_001191617.1
PLC
AAA28820.1
47%; 65%; (2x10-65)
38%; 56%; (4x10-53)
C/EBP
C/EBP gamma
AAC50201.1
ApC/EBP
NP_001191392.1
C/EBP
AAA28415.1
45%; 72%; (86x10-19)
40%; 73%; (4x10-15)
C/EBP delta
EAW86679.1
ApC/EBP
NP_001191392.1
C/EBP
AAA28415.1
38%; 69%; (1x10-14)
40%; 70%; (2x10-16)
C/EBP beta
EAW75629.1
ApC/EBP
NP_001191392.1
C/EBP
AAA28415.1
45%; 63%;(4x10-18)
44%; 68%; (1x10-16)
C/EBP épsilon
EAW66183.1
ApC/EBP
NP_001191392.1
C/EBP
AAA28415.1
30%; 50%; (6x10-17)
43%; 64%; (1x10-15)
C/EBP zeta
AAH34475.1
ApC/EBP
NP_001191392.1
C/EBP
AAA28415.1
29%;40%; (2.6)
34%; 48%; (0.078)
CaMKII-
CaMKII-
AAH40457.1
CaMKII-
ACN43221.1
CaM-kinase II alpha
Q00168.1
78%; 88%; (0.0)
71%; 81%; (0.0)
CN
CN
AAC37581.1
CN B homologous protein 1-like
XP_005095982.1
CN A1,
isoform A (D)
AAF57105.3
26%; 51%; (0.90)
24%; 48%; (1.8)
CN
AC37581.1
CN B,
isoform A (B)
AAF46026.1
36%; 50%; (4.6)
CPEB
CPEB1
Q9BZB8.1
CPEB
NP_001191467.1
orb2, isoform A (B,C,D, H)
AAF50352.1
65%;80%; (3x10-143)
42%; 59%; (2x10-63)
Table 2: Comparison of protein sequences involved in learning and memory by basic local alignment search tool (BLAST) in human (Homo sapiens), snail (Aplysia californica), and fly (Drosophila melanogaster). Abbreviations: AC: Adenylate Cyclase; AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid Receptor; C/EBP: CCAAT-box-enhanced Binding Protein; CaMKII: Ca2+/Calmodulin Protein Kinase II; CaMAC: Ca2+/calmodulin-activated Adenylyl Cyclase; Ca-i-PKC: Ca2+-independent PKC; CN: Calcineurin; CPEB: Cytoplasmic Polyadenylation Element Binding Protein; DAR: Dopamine Receptor; PLC: Phospholipase C; PP1: Protein Phosphatase-1; PKA: Protein Kinase A; PKC:Protein Kinase C; NMDAR: N-methyl-D-aspartate Receptor; UH: Ubiquitin Hydroxylase; 5-HT-R: Serotonin Receptor
![]()
CPEB2,3,4
Q7Z5Q1.3
CPEB
NP_001191467.1
orb2, isoform A
(B,C,D,H)
AAF50352.1
39%; 57%; (2x10-53)
88%; 93%; (2x10-174)
NMDA
AAA21180.1
NMDA-typeglutamate
receptor
precursor
NP_001191411.1
dNR 1
AAF52016.1
56%;67%; (0.0)
48%; 67%; (0.0)
NMDA
AAA21180.1
dNR 2, isoform A
(B,C,D,E,F,G)
AAN09051.2
29%; 47%; (3x10-91)
AMPA1
P42261.2
GluR1
AAP41203.1
glutamate receptor IA
AAF50652.2
40%; 57%; (2x10-170)
40%; 57%; (0.0)
AMPA1
P42261.2
glutamate receptor IB,
isoform A (B,C)
AAF50306.2
36%; 53%; (2x10-114)
AMPA2
P42262.3
GluR2
AAP41204.1
glutamate receptor IA
AAF50652.2
43%, 60%, (0.0)
42%; 58%; (0.0)
AMPA2
P42262.3
glutamate receptor IB
, isoform A (B,C)
AGB94317.1
36%; 53%; (2x10-114)
AMPA3
P42263.2
GluR3
AAP41205.1
glutamate receptor IA
AAF50652.2
44%; 62%; (0.0)
42%; 58%; (0.0)
AMPA3
P42263.2
glutamate receptor IB
, isoform A (B,C)
AAF50306.2
36%; 52%; (2x10-120)
AMPA4
P48058.2
GluR4
AAP41206.1
glutamate receptor IA
AAF50652.2
39%; 55%; (0.0)
43%; 60%; (0.0)
AMPA4
P48058.2
glutamate receptor IB, isoform A (B,C)
AAF50306.2
42%; 56%; (0.0)
5-HT-R
CAA05851.1
5-HT-R
ACJ63459.1
5-HT-R1
P20905.1
69%;84%;(0.45)
83%; 100%; (1.5)
5-HT-R
AAA66493.1
5-HT-R
ACJ63459.1
5-HT-R2A
P28285.2
39%; 58%; (3x10-93)
48%; 69%; (3x10-61)
5-HT-R
AAA66493.1
5-HT-R2B
P28286.3
48%; 68%; (3x10-62)
DAR D1A
P21728.1
DAR 1-like
XP_005100402.1
DAR
AAC47161.1
40%; 54%; (2x10-76)
35%; 48%; (6x10-59)
DAR D1B(5)
CAA41360.1
DAR1-like
XP_005100402.1
DAR
AAC47161.1
42%; 54%, (8x101-70)
42%;57%; (4x10-40)
DARD2
P14416.2
DAR1-like
XP_005100402.1
DAR
AAC47161.1
34%; 52%; (1x10-33)
34%; 49%; (1x10-62)
DARD3
P35462.2
DAR1-like
XP_005100402.1
DAR
AAC47161.1
32%; 49%; (8x10-43)
36%; 50%; (3x10-65)
DARD4
P21917.2
DAR1-like
XP_005100402.1
DAR
AAC47161.1
44%; 62%; (2x10-21)
40%; 54%; (8x10-26)
DAR D1A
P21728.1
DAR2-like
XP_005099999.1
DAR
CAA54451.1
40%; 57%; (2x10-37)
40%; 55%; (6x10-71)
DAR D1B(5)
CAA41360.1
DAR 2-like
XP_005099999.1
DAR
CAA54451.1
38%;53%; (8x10-36)
36%; 52%; (3x10-70)
DARD2
P14416.2
DAR 2-like
XP_005099999.1
DAR
CAA54451.1
32%; 49%; (2x10-63)
36%; 56%; (2x10-35)
DARD3
P35462.2
DAR2-like
XP_005099999.1
DAR
CAA54451.1
34%;49%; (1x10-58)
33%; 46%; (3x10-52)
DARD4
P21917.2
DAR2-like
XP_005099999.1
DAR
CAA54451.1
37%;54%; (3x10-25)
38%; 57%; (6x10-24)
Table 2 to 2:
Furthermore, NMR analysis of the prion-like N-terminal domain of CPEB has shown that, apart from beta-sheet, it also has helical and random-coil structures, which are essential for enhanced binding of mRNA-binding. These results support the notion that CPEB can act as a self-sustaining prion-like protein in the nervous system [102], thereby allowing the activity-dependent change in synaptic efficacy to persist for long periods of time. Similar to Aplysia, orthologues of CPEB have been reported in Drosophila (Orb2, [103]), mice (mCPEB3, [104]), and humans (hCPEB3, [105]).
Last, but not least, Barco and co-workers went further to show that striatum-based implicit memory and hippocampus-based explicit memory (e.g., spatial memory) in mice (and probably in humans) share common molecular mechanisms [106]. However, in contrast to sensitization and classical conditioning, spatial navigation involves the formation of a cognitive map in the hippocampus and entorhinal cortex modulated by “place cells” [17,107] and “grid cells”, respectively [108]. Indeed, these cells are involved in the position, direction, and distance information for accurate spatial navigation [109]. Since the formation of spatial navigation is a learning process [110], it requires the same molecular mechanism for the consolidation of implicit and explicit long term-memory. Amazingly, the fly Drosophila melanogaster not only displays olfactory memory (implicit), but also a visual place learning [111]. Given the deep homology between the fly and mammalian brain structures and neurobiology [112-114], we anticipate that “place cells” and “grid cells” might be located in the insect central complex –e.g., ellipsoid body [111], the homolog structure of mammalian hippocampus. This assumption suggests that implicit and “explicit” memory can be recreated on the fly due to distinct neuroanatomical substrates for spatial (e.g., [115]) versus non-spatial learning (e.g., mushroom bodies are equivalent to cerebellum [116], fan-shaped body to striatum [114]. Recently, Si’s laboratory has shown a protein network comprised of protein phosphatase 2A (PP2A), Transducer of Erb-B2 (Tob), and Lim Kinase (LimK) that controls the abundance of Orb2A [117]. Indeed, the interplay between PP2A and Tob-LimK activity may dynamically regulate Orb2 amyloid-like oligomer formation and the stabilization of memories in D. melanogaster.
Together, these findings demonstrated that implicit and explicit memory processes are not restricted to vertebrates. This suggests that there might be a unified molecular mechanism for the formation of stable long-term memories and synaptic plasticity in most sub-groups of Animalia (i.e., from vertebrates (mice, humans) to molluscs (snails: A. californica) to arthropods (insects: D. melanogaster) to annelids (Caenorhabditis elegans, [118]) to handle memory encoding and retrieval. Therefore, we think that the ease of genetic manipulation in D. melanogaster [119] and its validation as a model, not only for learning and memory [120-122], but also for neurodegeneration in AD [123], provides a unique opportunity to forge an integrated mechanistic understanding of memory processing and its alterations [124].
The gray side of memory
Given that implicit and explicit memory share common molecules that are evolutionarily conserved (Table 2), [125]), what are the physiological mechanisms responsible for memory persistence in HSAM cases? What are the molecular changes that explain the early stage (pre-mild cognitive impairment or pre-clinical) in familial AD (FAD)? Can the nature of both neurologic disorders stem from similar mechanisms, but in opposite directions?
Although no data are reasonably available to answer these questions, we propose that HSAM and FAD PSEN-1 E280A [126,127] are extreme, but opposing cases of the memory process. This assumption is based on the following observations. (i) The same morphological brain structures are mostly affected in both HSAM and FAD-PSEN-1, (e.g., the medial temporal lobe). In fact, while unusual expanded regions, such as the inferior and middle temporal gyri and temporal pole, the parahippocampal gyrus, and uncinate fascicle, have been detected in HSAM individuals [61], early hippocampus and cortical atrophy have been detected in FAD patients [128]. These observations suggest that those anatomical changes represent the strength of synaptic connections in HSAM, whereas loss of neuronal synaptic connections is progressively attained in FAD. Consequently, episodic/semantic memory and cognitive functions are highly retained in HSAM [61,129], but dramatically diminished in FAD [130-134], which are detected as measurable cognitive impairment around two decades before dementia onset in PSEN1 E280A carriers [135]. (ii) Both disorders present early age at onset. In fact, onset appears at about 10 years of age in HSAM patients compared to normal individuals [60] and about 37-45 years of age in FAD (i.e., range of dementia onset in FAD compared to the mean age of MCI onset of 45 years, [50,135]). (iii) Although not yet proved in HSAM individuals, these disorders might be generated either by gene mutations (e.g., presenilin-1 in FAD [136]), up-regulation (in HSAM) and/or down-regulation (in FAD)of the PKA/CREB-1/CPEB axis [91,93,94], alterations of synaptic calcium signaling [137,138], or altered metabolism of the amyloid beta precursor protein [139], which results in the over-generation of the peptide fragment Aβ1-42 [140] and aggregated Aβ as early as 28 years of age in asymptomatic PSEN-1 E280A mutation carriers [141]. Consequently, the molecular mechanism of consolidation and maintenance of memory at the synapse might be either up-regulated or down-regulated depending on the genetic make-up of each brain disorder.
On the understanding that oligomers of Aβ1-42 is the primary culprit of AD/FAD [142], and that Aβ1-42 amyloidogenesis can be initiated within living neurons rather than in the extracellular space [143-146], Aβ emerges as a molecule with a variety of cytotoxic effects. Effectively, it has been shown that Aβ is able to mediate mitochondrial toxicity [147], produce intracellular reactive oxygen species and alter calcium homeostasis [148], disrupt synaptic plasticity by altering glutamate recycling at the synapse [149], decrease spine density [150], impair the synaptic plasticity [151], and provoke neuronal loss [152] with disruption of memory-related synapse function [153]. Furthermore, Barucker and colleagues have recently shown that Aβ1-42 might act as a regulator of gene transcription [154].
Taken together, these data suggest that, though Aβ is a multifaceted peptide that can trigger different mechanisms of toxicity, little is known about the biological activities of Aβ [155,156]. Consequently, recurrent therapeutic failure (e.g., [157]) has paved the way to cast serious doubts on its role as a principal molecule in AD [158,159]. Therefore, further alternative ways in which Aβ operates might be explored (Karran and Hardy, 2014b [157]) before dismissing the amyloid cascade hypothesis (Hardy and Higgins, 1992 [169]).
We propose an intracellular amyloid-dependent mechanism at the neuronal synapse as the cause of early onset of FAD in which Aβ directly intermingles with the CPEB. As a result of “yin-yang” prionlike protein interactions [102], Aβ might be capable of interfering with CPEB’s normal function (i.e., control of long-term synaptic plasticity and maintenance of memory consolidation), thereby triggering synaptic apoptosis [160,161]. Therefore, synapse deterioration may lead to early age onset of AD (Figure 3B). Clearly, the toxic effect of oligomer Aβ might be mediated by an intracellular mechanism (e.g., [162,163]. Our assumptions are based on the evidence that Aβ oligomers are able to interact with prion protein and provoke cellular toxicity. Indeed, Peters et al. have recently found that Aβ interacts with the PrPc, a glycosylphosphatidylinositol (GPI)-anchored membrane prion protein, which functions as a receptor of Aβ1-42, to induce neuronal membrane damage and synaptotoxicity [164]. Dohler et al. have shown that, in Alzheimer’s disease brains, binding of Aβ to PrPc occurred via the PrPc N-terminus, which is important for proper Aβ-PrPc binding [165]. Hyeon et al. have shown that cells with abundant PrPc expression seemed to be more susceptible to Aβ1-42 oligomer toxicity [166].
If validated, our hypothesis may help explain why anti-amyloid therapies i.e., vaccines based on anti-Aβ antibodies [167,168] have been negative or inconclusive so far [158,169,170]. Therefore, efforts should be re-directed to design therapies that target intracellular Aβ oligomers. Alternatively, studies on HSAM individuals might be invaluable to discover which molecules can be up-regulated (e.g., PKA, CREB-1, CPEB) and/or down-regulated (e.g., CREB-2, Figure 3C) to increase memory abilities in FAD. Further research is warranted to confirm or dismiss such a thesis.
Acknowledgment
This work was sponsored by CODI (project EO 1617), Universidad de Antioquia (UdeA). We thank Hector Ortega-Arellano for preparing Table 2. Alejandra Ortiz-Ramirez is acknowledged for assistance with scientific literature and fruitful academic discussions.Her participation was a partial requirement for her training under the “Young Scientists Program” at the UdeA (contract #8700-216-2013).
References
- Ebbinghaus H. Memory: A Contribution to Experimental Psychology. In: Classics in Psychology, 1855-1914: Historical Essays. Bristol, UK: Thoemmes Press. 1999. Translated by Henry A. Ruger & Clara E. Bussenius (1913). Originally published in New York by Teachers College, Columbia University.
- Miller GA. The magical number seven plus or minus two: some limits on our capacity for processing information. Psychol Rev. 1956; 63: 81-97.
- Tulving E. Episodic memory: from mind to brain. Annu Rev Psychol. 2002; 53: 1-25.
- Curran T, Schacter DL. Implicit memory: what must theories of amnesia explain? Memory. 1997; 5: 37-47.
- Tulving E, Schacter DL. Priming and human memory systems. Science. 1990; 247: 301-306.
- Atkinson R, Shiffrin R. Human memory: A proposed system and its control processes. In: The psychology of learning and motivation. Spence KW SJ, editor. New York, Academic Press. 1968; 89-195.
- Neha, Sodhi RK, Jaggi AS, Singh N. Animal models of dementia and cognitive dysfunction. Life Sci. 2014; 109: 73-86.
- Papassotiropoulos A, Gerhards C, Heck A, Ackermann S, Aerni A, Schicktanz N, et al. Human genome-guided identification of memory-modulating drugs. Proc Natl Acad Sci U S A. 2013; 110: E4369-4374.
- Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry. 1957; 20: 11-21.
- Annese J, Schenker-Ahmed NM, Bartsch H, Maechler P, Sheh C, Thomas N, et al. Postmortem examination of patient H.M.'s brain based on histological sectioning and digital 3D reconstruction. Nat Commun. 2014; 5: 3122.
- Augustinack JC, van der Kouwe AJ, Salat DH, Benner T, Stevens AA, Annese J, et al. H.M.'s contributions to neuroscience: a review and autopsy studies. Hippocampus. 2014; 24: 1267-1286.
- Milner B. Disorders of learning and memory after temporal lobe lesions in man. Clin Neurosurg. 1972; 19: 421-446.
- Corkin S. What's new with the amnesic patient H.M.? Nat Rev Neurosci. 2002; 3: 153-160.
- Squire LR, Wixted JT. The cognitive neuroscience of human memory since H.M. Annu Rev Neurosci. 2011; 34: 259-288.
- Saksida LM, Bussey TJ. Animal models of amnesia. Neuropsychologia. 2010; 48: 2231-2233.
- Kandel ER. Nobel Lecture: The Molecular Biology of Memory Storage: A Dialog between Genes and Synapses". Nobelprize.org. Nobel Media AB. 2014.
- O'Keefe J. "Facts". Nobelprize.org. Nobel Media AB. 2014.
- Moser M-B. Nobel Lecture: Grid Cells, Place Cells and Memory". Nobelprize.org. Nobel Media AB. 2014.
- Moser EI. Facts. Nobelprize.org. Nobel Media AB. 2014.
- Milner B, Squire LR, Kandel ER. Cognitive neuroscience and the study of memory. Neuron. 1998; 20: 445-468.
- Corkin S. Permanent Present Tense: The Unforgettable Life of the Amnesic Patient, H. M.: Basic Books, a member of the Perseus Books Group. 2013.
- Wilson BA, Baddeley AD, Kapur N. Dense amnesia in a professional musician following herpes simplex virus encephalitis. J Clin Exp Neuropsychol. 1995; 17: 668-681.
- Wilson B, Wearing D. Prisoner of consciousness: A state of just awakening following herpes simplex encephalitis. In: Broken memories: Case studies in memory impairment. Campbell R, Conway MA, editors. Malden: Blackwell Publishing. 1995; 14-30.
- Wearing D. The man who keeps falling in love with his wife. The telegraph. 2005.
- Sacks O. The abyss. Music and amnesia. 2007.
- Alzheimer A. Ueber eine eigenartige Erkrankung der Hirnrinde [About a peculiar disease of the cerebral cortex]. 37th Meeting of psychiatrists of Sothwestern Germany in Tubingen on November 3rd and 4th 1906. Translation by C Schwab. Neuro Science News. 2000; 3: 4-7.
- Maurer K, Volk S, Gerbaldo H. Auguste D and Alzheimer's disease. Lancet. 1997; 349: 1546-1549.
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984; 120: 885-890.
- Müller U, Winter P, Graeber MB. A presenilin 1 mutation in the first case of Alzheimer's disease. Lancet Neurol. 2013; 12: 129-130.
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011; 7: 280-292.
- Weintraub S, Wicklund AH, Salmon DP. The neuropsychological profile of Alzheimer disease. Cold Spring Harb Perspect Med. 2012; 2: a006171.
- Jack CR Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009; 132: 1355-1365.
- Esiri M. The neuropathology of Alzheimer’s disease. In: Neurobiology of Alzheimer’s disease. Dawbarn D, SJ A, editors. Oxfrod University Press. 2001; 33-53.
- Braskie MN, Thompson PM. Understanding cognitive deficits in Alzheimer's disease based on neuroimaging findings. Trends Cogn Sci. 2013; 17: 510-516.
- Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013; 12: 207-216.
- Llorens-Martín M, Blazquez-Llorca L, Benavides-Piccione R, Rabano A, Hernandez F, Avila J, et al. Selective alterations of neurons and circuits related to early memory loss in Alzheimer's disease. Front Neuroanat. 2014; 8: 38.
- Tondelli M, Wilcock GK, Nichelli P, De Jager CA, Jenkinson M, Zamboni G. Structural MRI changes detectable up to ten years before clinical Alzheimer's disease. Neurobiol Aging. 2012; 33: 825.
- Spulber G, Niskanen E, Macdonald S, Kivipelto M, Padilla DF, Julkunen V, et al. Evolution of global and local grey matter atrophy on serial MRI scans during the progression from MCI to AD. Curr Alzheimer Res. 2012; 9: 516-524.
- Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, et al. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer's disease. Nat Neurosci. 2014; 17: 304-311.
- Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010; 9: 119-128.
- Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC, et al. The Alzheimer's Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement. 2013; 9: e111-194.
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002; 297: 353-356.
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991; 82: 239-259.
- de Toledo-Morrell L, Goncharova I, Dickerson B, Wilson RS, Bennett DA. From healthy aging to early Alzheimer's disease: in vivo detection of entorhinal cortex atrophy. Ann N Y Acad Sci. 2000; 911: 240-253.
- Devanand DP, Bansal R, Liu J, Hao X, Pradhaban G, Peterson BS. MRI hippocampal and entorhinal cortex mapping in predicting conversion to Alzheimer's disease. Neuroimage. 2012; 60: 1622-1629.
- Yokoyama S, Kajiya Y, Yoshinaga T, Tani A, Hirano H. Imaging discrepancies between magnetic resonance imaging and brain perfusion single-photon emission computed tomography in the diagnosis of Alzheimer's disease, and verification with amyloid positron emission tomography. Psychogeriatrics. 2014; 14: 110-117.
- Frisoni GB, Bocchetta M, Chételat G, Rabinovici GD, de Leon MJ, Kaye J, et al. Imaging markers for Alzheimer disease: which vs how. Neurology. 2013; 81: 487-500.
- Sperling RA, Karlawish J, Johnson KA. Preclinical Alzheimer disease-the challenges ahead. Nat Rev Neurol. 2013; 9: 54-58.
- Lalli MA, Cox HC, Arcila ML, Cadavid L, Moreno S, Garcia G, et al. Origin of the PSEN1 E280A mutation causing early-onset Alzheimer's disease. Alzheimers Dement. 2014; 10: S277-277S283.
- Kosik KS, Muñoz C, Lopez L, Arcila ML, García G, Madrigal L, et al. Homozygosity of the autosomal dominant Alzheimer disease presenilin 1 E280A mutation. Neurology. 2015; 84: 206-208.
- Rosenbaum RS, Gilboa A, Moscovitch M. Case studies continue to illuminate the cognitive neuroscience of memory. Ann N Y Acad Sci. 2014; 1316: 105-133.
- Ranganath C, Blumenfeld RS. Doubts about double dissociations between short- and long-term memory. Trends Cogn Sci. 2005; 9: 374-380.
- Graham KS, Barense MD, Lee AC. Going beyond LTM in the MTL: a synthesis of neuropsychological and neuroimaging findings on the role of the medial temporal lobe in memory and perception. Neuropsychologia. 2010; 48: 831-853.
- Jeneson A, Squire LR. Working memory, long-term memory, and medial temporal lobe function. Learn Mem. 2011; 19: 15-25.
- Kim S, Sapiurka M, Clark RE, Squire LR. Contrasting effects on path integration after hippocampal damage in humans and rats. Proc Natl Acad Sci U S A. 2013; 110: 4732-4737.
- Smith CN, Jeneson A, Frascino JC, Kirwan CB, Hopkins RO, Squire LR. When recognition memory is independent of hippocampal function. Proc Natl Acad Sci U S A. 2014; 111: 9935-9940.
- Smith CN, Urgolites ZJ, Hopkins RO, Squire LR. Comparison of explicit and incidental learning strategies in memory-impaired patients. Proc Natl Acad Sci U S A. 2014; 111: 475-479.
- Smith CN, Frascino JC, Hopkins RO, Squire LR. The nature of anterograde and retrograde memory impairment after damage to the medial temporal lobe. Neuropsychologia. 2013; 51: 2709-2714.
- Price J, Davis B. The Woman Who Can't Forget: The Extraordinary Story of Living with the Most Remarkable Memory Known to Science. A Memoir. Free Press 2008.
- Parker ES, Cahill L, McGaugh JL. A case of unusual autobiographical remembering. Neurocase. 2006; 12: 35-49.
- LePort AK, Mattfeld AT, Dickinson-Anson H, Fallon JH, Stark CE, Kruggel F, et al. Behavioral and neuroanatomical investigation of Highly Superior Autobiographical Memory (HSAM). Neurobiol Learn Mem. 2012; 98: 78-92.
- Martin M, Clare L, Altgassen AM, Cameron MH, Zehnder F. Cognition-based interventions for healthy older people and people with mild cognitive impairment. Cochrane Database Syst Rev. 2011; : CD006220.
- Bryck RL, Fisher PA. Training the brain: practical applications of neural plasticity from the intersection of cognitive neuroscience, developmental psychology, and prevention science. Am Psychol. 2012; 67: 87-100.
- Kueider AM, Parisi JM, Gross AL, Rebok GW. Computerized cognitive training with older adults: a systematic review. PLoS One. 2012; 7: e40588.
- Treffert DA. The savant syndrome: an extraordinary condition. A synopsis: past, present, future. Philos Trans R Soc Lond B Biol Sci. 2009; 364: 1351-1357.
- Treffert DA, Christensen DD. Inside the mind of a savant. Sci Am. 2005; 293: 108-113.
- Peek K. The Real Rain Man" (Documentary). Focus Productions, Bristol, England, UK. 2006.
- Peek F, Hanson L. The Life and Message of the Real Rain Man: The Journey of a Mega-Savant. Dute Publishing. 2008.
- Corrigan NM, Richards TL, Treffert DA, Dager SR. Toward a better understanding of the savant brain. Compr Psychiatry. 2012; 53: 706-717.
- Treffert DA. Savant syndrome: realities, myths and misconceptions. J Autism Dev Disord. 2014; 44: 564-571.
- Chi RP, Snyder AW. Brain stimulation enables the solution of an inherently difficult problem. Neurosci Lett. 2012; 515: 121-124.
- Snyder A. Explaining and inducing savant skills: privileged access to lower level, less-processed information. Philos Trans R Soc Lond B Biol Sci. 2009; 364: 1399-1405.
- Gobet F, Snyder A, Bossomaier T, Harré M. Designing a "better" brain: insights from experts and savants. Front Psychol. 2014; 5: 470.
- Opitz JM, Smith JF, Santoro L. The FG syndromes (Online Mendelian Inheritance in Man 305450): perspective in 2008. Adv Pediatr. 2008; 55: 123-170.
- Opitz JM, Kaveggia EG. Studies of malformation syndromes of man 33: the FG syndrome. An X-linked recessive syndrome of multiple congenital anomalies and mental retardation. Z Kinderheilkd. 1974; 117: 1-18.
- Thompson E, Baraitser M. FG syndrome. J Med Genet. 1987; 24: 139-143.
- Svoboda E, McKinnon MC, Levine B. The functional neuroanatomy of autobiographical memory: a meta-analysis. Neuropsychologia. 2006; 44: 2189-2208.
- Hall SS. The quest for a smart pill. Sci Am. 2003; 289: 54-57, 60-5.
- Pavlopoulos E, Jones S, Kosmidis S, Close M, Kim C, Kovalerchik O, et al. Molecular mechanism for age-related memory loss: the histone-binding protein RbAp48. Sci Transl Med. 2013; 5: 200ra115.
- Kandel E, Siegelbaum S. Cellular mechanisms of implicit memory storage and the biological basis of individuality. In: Principles of Neural Science, 5th edn. Kandel ER SH, Jessell TM, Siegelbaum SA, Hudspeth AJ, editor. New York McGraw-Hill. 2012; 1461-186.
- Kandel ER. Nobel Lecture: The Molecular Biology of Memory Storage: A Dialog between Genes and Synapses. Nobelprize.org. Nobel Media AB. 2014.
- Barco A, Pittenger C, Kandel ER. CREB, memory enhancement and the treatment of memory disorders: promises, pitfalls and prospects. Expert Opin Ther Targets. 2003; 7: 101-114.
- Hawkins RD, Kandel ER, Bailey CH. Molecular mechanisms of memory storage in Aplysia. Biol Bull. 2006; 210: 174-191.
- Bailey CH, Kandel ER. Synaptic remodeling, synaptic growth and the storage of long-term memory in Aplysia. Prog Brain Res. 2008; 169: 179-198.
- Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012; 5: 14.
- Mayford M, Siegelbaum SA, Kandel ER. Synapses and memory storage. Cold Spring Harb Perspect Biol. 2012; 4.
- Alberini CM, Kandel ER. The regulation of transcription in memory consolidation. Cold Spring Harb Perspect Biol. 2014; 7: a021741.
- Kandel ER, Dudai Y, Mayford MR. The molecular and systems biology of memory. Cell. 2014; 157: 163-186.
- Kandel E. In Search of Memory: The Emergence of a New Science of Mind. New York. 2007.
- Castellucci VF, Carew TJ, Kandel ER. Cellular analysis of long-term habituation of the gill-withdrawal reflex of Aplysia californica. Science. 1978; 202: 1306-1308.
- Suzuki A, Fukushima H, Mukawa T, Toyoda H, Wu LJ, Zhao MG, et al. Upregulation of CREB-mediated transcription enhances both short- and long-term memory. J Neurosci. 2011; 31: 8786-8802.
- Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002; 108: 689-703.
- Restivo L, Tafi E, Ammassari-Teule M, Marie H. Viral-mediated expression of a constitutively active form of CREB in hippocampal neurons increases memory. Hippocampus. 2009; 19: 228-234.
- Marchetti C, Tafi E, Marie H. Viral-mediated expression of a constitutively active form of cAMP response element binding protein in the dentate gyrus increases long term synaptic plasticity. Neuroscience. 2011; 190: 21-26.
- Yin JC, De Vecchio M, Zhou H, Tully T. CREB as a memory modulator: induced expression of a dCREB2 activator isoform enhances long-term memory in Drosophila. Cell. 1995; 81: 107-115.
- Chen A, Muzzio IA, Malleret G, Bartsch D, Verbitsky M, Pavlidis P, et al. Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB-2) and C/EBP proteins. Neuron. 2003; 39: 655-669.
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, et al. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem. 2005; 12: 111-119.
- Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, et al. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell. 2003; 115: 893-904.
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell. 2003; 115: 879-891.
- Si K, Choi YB, White-Grindley E, Majumdar A, Kandel ER. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell. 2010; 140: 421-435.
- Raveendra BL, Siemer AB, Puthanveettil SV, Hendrickson WA, Kandel ER, McDermott AE, et al. Characterization of prion-like conformational changes of the neuronal isoform of Aplysia CPEB. Nat Struct Mol Biol. 2013; 20: 495-501.
- Charlesworth A, Meijer HA, de Moor CH. Specificity factors in cytoplasmic polyadenylation. Wiley Interdiscip Rev RNA. 2013; 4: 437-461.
- Majumdar A, Cesario WC, White-Grindley E, Jiang H, Ren F, Khan MR, et al. Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell. 2012; 148: 515-529.
- Pavlopoulos E, Trifilieff P, Chevaleyre V, Fioriti L, Zairis S, Pagano A, et al. Neuralized1 activates CPEB3: a function for nonproteolytic ubiquitin in synaptic plasticity and memory storage. Cell. 2011; 147: 1369-1383.
- Vogler C, Spalek K, Aerni A, Demougin P, Müller A, Huynh KD, et al. CPEB3 is associated with human episodic memory. Front Behav Neurosci. 2009; 3: 4.
- Barco A, Bailey CH, Kandel ER. Common molecular mechanisms in explicit and implicit memory. J Neurochem. 2006; 97: 1520-1533.
- O'Keefe J. A review of the hippocampal place cells. Prog Neurobiol. 1979; 13: 419-439.
- Fyhn M, Hafting T, Witter MP, Moser EI, Moser MB. Grid cells in mice. Hippocampus. 2008; 18: 1230-1238.
- Moser E, Moser MB. Mapping your every move. Cerebrum. 2014; 2014: 4.
- Muzzio IA, Kentros C, Kandel E. What is remembered? Role of attention on the encoding and retrieval of hippocampal representations. J Physiol. 2009; 587: 2837-2854.
- Ofstad TA, Zuker CS, Reiser MB. Visual place learning in Drosophila melanogaster. Nature. 2011; 474: 204-207.
- Nichols CD. Drosophila melanogaster neurobiology, neuropharmacology, and how the fly can inform central nervous system drug discovery. Pharmacol Ther. 2006; 112: 677-700.
- Farris SM. Are mushroom bodies cerebellum-like structures? Arthropod Struct Dev. 2011; 40: 368-379.
- Strausfeld NJ, Hirth F. Deep homology of arthropod central complex and vertebrate basal ganglia. Science. 2013; 340: 157-161.
- Foucaud J, Burns JG, Mery F. Use of spatial information and search strategies in a water maze analog in Drosophila melanogaster. PLoS One. 2010; 5: e15231.
- Busto GU, Cervantes-Sandoval I, Davis RL. Olfactory learning in Drosophila. Physiology (Bethesda). 2010; 25: 338-346.
- White-Grindley E, Li L, Mohammad Khan R, Ren F, Saraf A, Florens L, et al. Contribution of Orb2A stability in regulated amyloid-like oligomerization of Drosophila Orb2. PLoS Biol. 2014; 12: e1001786.
- Stein GM, Murphy CT. C. elegans positive olfactory associative memory is a molecularly conserved behavioral paradigm. Neurobiol Learn Mem. 2014; 115: 86-94.
- del Valle Rodríguez A, Didiano D, Desplan C. Power tools for gene expression and clonal analysis in Drosophila. Nat Methods. 2011; 9: 47-55.
- McGuire SE, Deshazer M, Davis RL. Thirty years of olfactory learning and memory research in Drosophila melanogaster. Prog Neurobiol. 2005; 76: 328-347.
- Perisse E, Burke C, Huetteroth W, Waddell S. Shocking revelations and saccharin sweetness in the study of Drosophila olfactory memory. Curr Biol. 2013; 23: R752-763.
- Das G, Klappenbach M, Vrontou E, Perisse E, Clark CM, Burke CJ, et al. Drosophila learn opposing components of a compound food stimulus. Curr Biol. 2014; 24: 1723-1730.
- Prüßing K, Voigt A, Schulz JB. Drosophila melanogaster as a model organism for Alzheimer's disease. Mol Neurodegener. 2013; 8: 35.
- Besnard A, Caboche J, Laroche S. Reconsolidation of memory: a decade of debate. Prog Neurobiol. 2012; 99: 61-80.
- Glanzman DL. Common mechanisms of synaptic plasticity in vertebrates and invertebrates. Curr Biol. 2010; 20: R31-36.
- Lopera F, Ardilla A, Martínez A, Madrigal L, Arango-Viana JC, Lemere CA, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997; 277: 793-799.
- Arcos-Burgos M, Muenke M. Genetics of population isolates. Clin Genet. 2002; 61: 233-247.
- Quiroz YT, Stern CE, Reiman EM, Brickhouse M, Ruiz A, Sperling RA, et al. Cortical atrophy in presymptomatic Alzheimer's disease presenilin 1 mutation carriers. J Neurol Neurosurg Psychiatry. 2013; 84: 556-561.
- McGaugh JL, LePort A. Remembrance of all things past. Sci Am. 2014; 310: 40-45.
- Cuetos F, Arango-Lasprilla JC, Uribe C, Valencia C, Lopera F. Linguistic changes in verbal expression: a preclinical marker of Alzheimer's disease. J Int Neuropsychol Soc. 2007; 13: 433-439.
- Arango-Lasprilla JC, Cuetos F, Valencia C, Uribe C, Lopera F. Cognitive changes in the preclinical phase of familial Alzheimer's disease. J Clin Exp Neuropsychol. 2007; 29: 892-900.
- Quiroz YT, Budson AE, Celone K, Ruiz A, Newmark R, Castrillón G, et al. Hippocampal hyperactivation in presymptomatic familial Alzheimer's disease. Ann Neurol. 2010; 68: 865-875.
- Quiroz YT, Ally BA, Celone K, McKeever J, Ruiz-Rizzo AL, Lopera F, et al. Event-related potential markers of brain changes in preclinical familial Alzheimer disease. Neurology. 2011; 77: 469-475.
- Parra MA, Sala SD, Abrahams S, Logie RH, Méndez LG, Lopera F, et al. Specific deficit of colour-colour short-term memory binding in sporadic and familial Alzheimer's disease. Neuropsychologia. 2011; 49: 1943-1952.
- Acosta-Baena N, Sepulveda-Falla D, Lopera-Gómez CM, Jaramillo-Elorza MC, Moreno S, Aguirre-Acevedo DC, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer's disease: a retrospective cohort study. Lancet Neurol. 2011; 10: 213-220.
- Alzheimer's Disease Collaborative Group. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genet. 1995; 11: 219-222.
- Lee SH, Sharma M, Südhof TC, Shen J. Synaptic function of nicastrin in hippocampal neurons. Proc Natl Acad Sci U S A. 2014; 111: 8973-8978.
- Sepulveda-Falla D, Barrera-Ocampo A, Hagel C, Korwitz A, Vinueza-Veloz MF, Zhou K, et al. Familial Alzheimer's disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J Clin Invest. 2014; 124: 1552-1567.
- Van Vickle GD, Esh CL, Kokjohn TA, Patton RL, Kalback WM, Luehrs DC, et al. Presenilin-1 280Glu-->Ala mutation alters C-terminal APP processing yielding longer abeta peptides: implications for Alzheimer's disease. Mol Med. 2008; 14: 184-194.
- Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, et al. The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat Med. 1996; 2: 1146-1150.
- Fleisher AS, Chen K, Quiroz YT, Jakimovich LJ, Gomez MG, Langois CM, et al. Florbetapir PET analysis of amyloid-β deposition in the presenilin 1 E280A autosomal dominant Alzheimer's disease kindred: a cross-sectional study. Lancet Neurol. 2012; 11: 1057-1065.
- Viola KL, Klein WL. Amyloid β oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015; 129: 183-206.
- Skovronsky DM, Doms RW, Lee VM. Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in culture. J Cell Biol. 1998; 141: 1031-1039.
- Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000; 39: 10831-10839.
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007; 8: 499-509.
- Gouras GK, Willén K, Tampellini D. Critical role of intraneuronal Aβ in Alzheimer's disease: technical challenges in studying intracellular Aβ. Life Sci. 2012; 91: 1153-1158.
- Kaminsky YG, Tikhonova LA, Kosenko EA. Critical analysis of Alzheimer's amyloid-beta toxicity to mitochondria. Front Biosci (Landmark Ed). 2015; 20: 173-197.
- Gray NE, Sampath H, Zweig JA, Quinn JF, Soumyanath A. Centella asiatica Attenuates Amyloid-β-Induced Oxidative Stress and Mitochondrial Dysfunction. J Alzheimers Dis. 2015; 45: 933-946.
- Varga E, Juhász G, Bozsó Z, Penke B, Fülöp L, Szegedi V, et al. Amyloid-β1-42 Disrupts Synaptic Plasticity by Altering Glutamate Recycling at the Synapse. J Alzheimers Dis. 2015; 45: 449-456.
- Borbély E, Horváth J, Furdan S, Bozsó Z, Penke B, Fülöp L. Simultaneous changes of spatial memory and spine density after intrahippocampal administration of fibrillar aß1-42 to the rat brain. Biomed Res Int. 2014; 2014: 345305.
- Sivanesan S, Tan A, Rajadas J. Pathogenesis of Abeta oligomers in synaptic failure. Curr Alzheimer Res. 2013; 10: 316-323.
- Feng J, Meng C, Xing D. Aβ induces PUMA activation: a new mechanism for Aβ-mediated neuronal apoptosis. Neurobiol Aging. 2015; 36: 789-800.
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002; 416: 535-539.
- Barucker C, Harmeier A, Weiske J, Fauler B, Albring KF, Prokop S, et al. Nuclear translocation uncovers the amyloid peptide Aβ42 as a regulator of gene transcription. J Biol Chem. 2014; 289: 20182-20191.
- Puzzo D, Privitera L, Palmeri A. Hormetic effect of amyloid-β peptide in synaptic plasticity and memory. Neurobiol Aging. 2012; 33: 1484.
- Morley JE, Farr SA. Hormesis and amyloid-β protein: physiology or pathology? J Alzheimers Dis. 2012; 29: 487-492.
- Karran E, Hardy J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol. 2014; 76: 185-205.
- Mullane K, Williams M. Alzheimer's therapeutics: continued clinical failures question the validity of the amyloid hypothesis-but what lies beyond? Biochem Pharmacol. 2013; 85: 289-305.
- Castello MA, Soriano S. On the origin of Alzheimer's disease. Trials and tribulations of the amyloid hypothesis. Ageing Res Rev. 2014; 13: 10-12.
- Costa-Mattioli M, Sonenberg N, Richter JD. Translational regulatory mechanisms in synaptic plasticity and memory storage. Prog Mol Biol Transl Sci. 2009; 90: 293-311.
- Mattson MP, Keller JN, Begley JG. Evidence for synaptic apoptosis. Exp Neurol. 1998; 153: 35-48.
- Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FA. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease. Neuroscience. 2005; 132: 123-135.
- Ripoli C, Cocco S, Li Puma DD, Piacentini R, Mastrodonato A, Scala F, et al. Intracellular accumulation of amyloid-β (Aβ) protein plays a major role in Aβ-induced alterations of glutamatergic synaptic transmission and plasticity. J Neurosci. 2014; 34: 12893-12903.
- Peters C, Espinoza MP, Gallegos S, Opazo C, Aguayo LG. Alzheimer's Aß interacts with cellular prion protein inducing neuronal membrane damage and synaptotoxicity. Neurobiol Aging. 2014.
- Dohler F, Sepulveda-Falla D, Krasemann S, Altmeppen H, Schlüter H, Hildebrand D, et al. High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer's disease. Brain. 2014; 137: 873-886.
- Hyeon JW, Kim SY, Park JS, Choi BY, Lee SM, Ju YR, et al. The association between prion proteins and Aβ₁₋₄₂‚ oligomers in cytotoxicity and apoptosis. Biochem Biophys Res Commun. 2012; 424: 214-220.
- Lobello K, Ryan JM, Liu E, Rippon G, Black R. Targeting Beta amyloid: a clinical review of immunotherapeutic approaches in Alzheimer's disease. Int J Alzheimers Dis. 2012; 2012: 628070.
- Galimberti D, Ghezzi L, Scarpini E. Immunotherapy against amyloid pathology in Alzheimer's disease. J Neurol Sci. 2013; 333: 50-54.
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992; 256: 184-185.
- Karran E, Hardy J. Antiamyloid therapy for Alzheimer's disease--are we on the right road? N Engl J Med. 2014; 370: 377-378.