
Case Report
Austin J Clin Neurol 2015;2(6): 1049.
Sturge-Weber Syndrome: A Case Report in a 39 Yr- Old Man with Delayed Diagnosis
MUSTAPHA Adekunle Fatai¹*, ADEBANJO Oluyemi Mary² and ADEBIMPE Oluwafisayo Adenike³
¹Department of Medicine, Division of Neurology, Ladoke Akintola University of Technology, Nigeria
²Department of Medicine, LAUTECH Teaching Hospital, Nigeria
³Department of Behavioural Sciences, LAUTECH Teaching Hospital, Nigeria
*Corresponding author: Mustapha Adekunle Fatai, Department of Medicine, Division of Neurology, Ladoke Akintola University of Technology (LAUTECH) Teaching Hospital, Osogbo Osun state, Nigeria
Received: April 07, 2015; Accepted: May 10, 2015; Published: May 17, 2015
Abstract
Sturge Weber Syndrome is a rare neurocutaneous disease characterized by facial port-wine stain, ocular abnormalities (glaucoma and choroidal haemangioma), and leptomeningeal angioma most often involving the occipital and posterior parietal lobes. This syndrome consists of a constellation of clinical features such as a facial neavus, seizures, hemiparesis, intracranial calcifications and mental retardation. We report a 39 year old cobbler who presented at the neurology clinic of LAUTECH Teaching Hospital Osogbo Nigeria on account of recurrent generalized seizures and learning difficulties since childhood. On examination, he had a right hyperpigmented periorbital patch with chemosis and complete visual loss in the right eye.
Electroencephalography (EEG) revealed generalized epileptiform activity and brain CT scan yielded multiple cerebral calcifications in keeping with Sturge Weber Syndrome. Patient is currently on carbamazepine, Folic acid and Cognitol and seizure control has improved remarkably and is being followed up at our out-patient clinic.
Keywords: Sturge Weber syndrome; Angioma; Seizure; Mental retardation
Introduction
Sturge-Weber syndrome, also known as encephalotrigerminal angiomatosis is a rare neurocutaneous disease characterized by facial port-wine stain, leptomeningeal angioma with or without glaucoma [1].
The Syndrome can be classified into 3 according to Roach Scale.
Type I – Facial and leptomeningeal angioma. Patient may have glaucoma.
Type II – Facial angioma only (without CNS involvement). Patient may have glaucoma
Type III – Leptomeningeal angioma only, usually without glaucoma.
Sturge Weber Syndrome (SWS) was first described by Schirmer in 1860 and later more comprehensively by Sturge in 1879, who associated dermatological and ophthalmological changes of the disease to the neurological manifestations. Weber complemented it with the documentation of radiologic findings seen in these patients [2]. It has a frequency of 1:50,000 live births [3]. It occurs sporadically all over the world and cuts across racial, gender and age divides. There are few hospital reports about this disease from Nigeria.
Case Presentation
A 39yr-old man was brought to the neurology clinic of LAUTECH Teaching Hospital Osogbo Nigeria by the mother in late 2012, on account of a history of recurrent generalized seizure and learning difficulties since childhood.
Seizure started before his first birthday, with frequency of one episode per day. The seizures were initially atonic but as he grew up, they became characterized by abnormal behavior such as being dazed, fidgeting with objects or running aimlessly around the house. These manifestations evolved over time to generalized tonic-clonic seizures preceded by loud cries and associated with tongue biting, foaming from the mouth and recurrent falls. The seizure frequency was about 2-3 per week and lasted about 2-3 minutes a few months prior to presentation and patient usually had postictal weakness and occasional urinary incontinence. He was treated with traditional medicine without much success. He suffered several burns injury as a result of the seizure as well as several episodes of tooth loss. He dropped out of school while in junior secondary school due to learning difficulty and poor academic performance. He has since found it difficult to learn a trade and has settled down to be a parttime shoe repairer. He had no history of hemiparesis, recurrent headaches or oral abnormalities of note, is the pregnancy history of the mother who took several unspecified drugs to abort the pregnancy that resulted into his birth owing to an existing large family size.
He was born with a port-wine stain around the right eye. The stain gradually became darker as he grew older. The right eye was also noticed to be bulging out as he was growing up and becoming red in the process. He lost vision in the same eye at age of 7.
On examination, he had a barely visible hyperpigmented patch in the distribution of ophthalmic branch of right trigeminal nerve as shown in Figure 1 and a nevus on the medial aspect of the right arm as seen in Figure 2. He also had several keloid scars on his trunk as depicted in Figure 3.
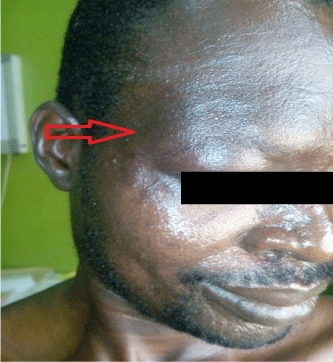
Figure 1: The arrow points to the faint hyperpigmented right periorbital patch
on the face of the patient.
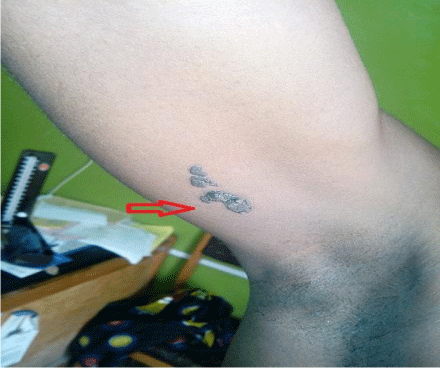
Figure 2: A neavus on the medial aspect of the right arm of the patient.
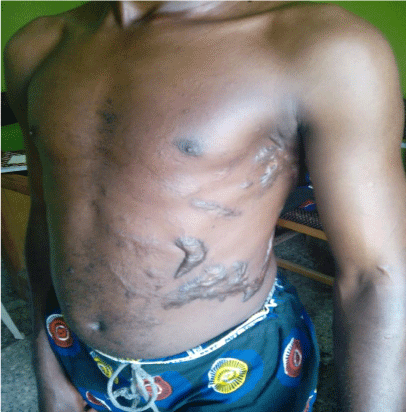
Figure 3: Several keloidal scars on the trunk of the patient as a result of scald
burns injuries.
Examination of the right eye revealed a visual acuity of no light perception, conjunctival chemosis, episcleral haemangioma with raised intraocular pressure. The fundus could not be viewed due to non dilating pupil and absent lens. Findings in the left eye were unremarkable.
Psychological examination showed he had an Intelligence quotient (IQ) of 14.4 using draw-a- person test.
EEG revealed generalized epileptiform activity.
Cranial CT scan showed multiple gyriform calcifications in the right occipital and parietal lobes, hyperpneumatization of right frontal sinus, and enlargement of right choroid plexus. The left cerebral hemisphere, midbrain and cerebellum were normal. This is illustrated in Figure 4.

Figure 4: Cranial CT scan showed multiple gyri form calcifications in the right
occipital and parietal lobes, hyperpneumatization of right frontal sinus, and
enlargement of right choroid plexus. The left cerebral hemisphere, midbrain
and cerebellum were normal.
He had blood tests such as electrolytes and Urea, fasting blood glucose, full blood count among others which were within normal limits.
A diagnosis of Sturge Weber Syndrome was made based on clinical features as well as the highly suggestive CT Brain findings. Patient was subsequently commenced on oral Carbamazepine, folic acid and Cognitol and seizure control has improved remarkably sometimes with no seizure within 1-2 months. He is currently being followed up at the Neurology clinic of LAUTECH Teaching Hospital Osogbo Nigeria.
Discussion
Sturge-Weber Syndrome is rare congenital neurocutaneous disorder which occurs sporadically. Only few cases have been reported in Nigeria; one reported in a three-month old infant by Baba Usman Ahmadu, et al. [4], another in a 56-yr old woman by Alli SK, et al [5].
SWS are caused by mutation of GNAQ gene (Guanine nucleotide binding protein subunit alpha gene) on chromosome 9q21 [6]. The reason for this mutation is not known but considering the fact that mother of our patient attempted to abort his pregnancy using several drugs, the possibility of a pharmacological agent causing SWS, if taken in pregnancy needs to be explored.
SWS presents with vascular malformations in the skin, eye and the brain. These malformations result from failure of normal regression of fetal vascular plexus surrounding the cephalic portion of the neural tube. The ectoderm over this area later forms the facial skin.
The typical presentations are port-wine stain on the face, neurological and ocular abnormalities. Our patient presented with all these features. He had a port-wine stain at birth though at presentation it was a hyperpigmented patch that was visible in the distribution of ophthalmic branch of right trigeminal nerve. He also had a naevus on the medial aspect of the right arm.
Port-wine stains in SWS are often found in the distribution of V1 and V2 branches of trigeminal nerve. In few cases, it may spread to the neck, chest and upper limbs. The stain may be difficult to appreciate in dark –skinned patients.
According to a study by Enjolras, et al. SWS occurs only when the nevus involves the V1 distribution of trigeminal nerve [7].
Common neurological manifestations of SWS are seizures, headaches, developmental delay, mental retardation, learning disorders, focal deficits like hemiparesis and hemianopsia.
These CNS features are caused by leptomeningeal angioma either as a result of mass effect on the brain or by causing chronic cerebral ischaemia via venous hypertension and vascular steal syndrome.
Our patient presented with seizure disorder and mental retardation. Seizure is seen in 75-90% of patients with SWS [8]. According to study done by Sturge Weber Foundation, median age of onset of seizure is six months [9].
According to Roach, early onset of seizure (before age 2) is associated with refractory epilepsy and mental retardation [10].
Seizure in our patient cannot be described as refractory because he was on traditional remedies for several years rather than conventional drugs. Seizure control improved remarkably after commencement of carbamazepine. However, long duration of uncontrolled seizure is a contributing factor to the degree of mental retardation seen in this patient.
Ocular features of SWS include glaucoma & vascular malformations of the conjunctiva, episclera, choroid and retina.
Glaucoma is the commonest eye symptom in SWS. It usually occurs only when there is ipsilateral facial PWS. Contralateral glaucoma is rare. It may be present at birth or can develop at any age. Incidence of glaucoma ranges between 30-71% of patients [10-12].
Sullivan et al. found glaucoma in 71% of the 51 patients with SWS reviewed for ocular abnormalities [12]. 69% had conjunctival or episcleral hemangiomas and 55% had choroidal hemangiomas [12].
According to data from Sturge-Weber Foundation, 48% of 171 patients studied had glaucoma [13].
Considering the blindness and raised intraocular pressure in the right eye of our patient, the possibility of glaucoma is very high.
Conclusion
This is a case of delayed diagnosis due to late presentation. SWS, though rare, can be diagnosed on clinical grounds. Early diagnosis and treatment prevents neurological deterioration.
References
- Thomas-Sohl KA, Vaslow DF, Maria BL. Sturge-Weber syndrome: a review. Pediatr Neurol. 2004; 30: 303-310.
- Neto FXP, Junior MAV, Ximanes LS, de Souza Jacob CC, Junior AGR, Paletha ACP. Clinical features of Sturge Weber Syndrome. Intl Arch Otorhinolaryngol. 2008; 12: 565-570.
- Welty LD. Sturge-Weber syndrome: A case study. Neonatal Netw. 2006; 25: 89-98.
- Baba Usman Ahmadu, Andrew Godiya, Murey Raphael Bartholomew, Ahmed Olayemi Bello. Sturge-Weber Syndrome in a three month old infant: a case report. International Research Journal of Basic and Clinical Studies. 2013; 1: 32-34.
- Alli SK, Adenuga OO, Ogbuagu MN, Velle LD, Akinyemi AO. Sturge-Weber syndrome in a 56 year old woman: a case report. Niger J Med. 2005; 14: 319-321.
- Kennedy Krieger Institute. “Game-changing discovery of gene mutation that causes Sturge-Weber syndrome, port-wine stain birthmarks offers new hope.” Science Daily. Science Daily. 2013.
- Enjolras O, Riche MC, Merland JJ. Facial port-wine stains and Sturge-Weber syndrome. Pediatrics. 1985; 76: 48-51.
- Kugler RM. Sturge-Weber syndrome affects skin and brain.
- Bodensteiner JB, Roach ES. Sturge –Weber Syndrome: Introduction and overview. In: Sturge-Weber Syndrome. Bodensteiner JB, Roach ES, editors. Sturge Weber Foundation, Mt. Freedom, NJ. 1999: 1-10.
- Roach ES, Bodensteiner JB. Neurologic manifestations of Sturge-Weber Syndrome. In: Sturge-Weber Syndrome. Mt Freedom, New Jersey: Sturge Weber Foundation; 1999: 27-38.
- Sullivan TJ, Clarke MP, Morin JD. The ocular manifestations of the Sturge-Weber syndrome. J Pediatr Ophthalmol Strabismus. 1992; 29: 349-356.
- Paller AS. The Sturge-Weber syndrome. Pediatr Dermatol. 1987; 4: 300-304.
- Takeota M, Riviello JJ. Sturge Weber Syndrome.