
Review Article
Austin J Clin Neurol 2015;2(7): 1060.
Unhealthy Diets Determine Benign or Toxic Amyloid Beta States and Promote Brain Amyloid Beta Aggregation
Martins IJ1,2,3*
¹Centre of Excellence in Alzheimer’s disease Research and Care, Edith Cowan University, Australia
²School of Psychiatry and Clinical Neurosciences, University of Western Australia, Australia
³McCusker Alzheimer’s Research Foundation, Hollywood Medical Centre, Australia
*Corresponding author: Ian Martins, School of Medical Sciences, Edith Cowan University, 270 Joondalup Drive, Joondalup, Western Australia 6027, Australia
Received: April 21, 2015; Accepted: June 26, 2015; Published: June 29, 2015
Abstract
Interests in amyloid beta oligomers and their relevance to mechanisms for toxic amyloid beta species has accelerated with effects on neuronal apoptosis in Alzheimer’s disease. Unhealthy diets that accelerate amyloidogenic pathways may involve lipids such as palmitic acid and cholesterol that promote hydrophobic self association reactions with amyloid beta aggregation in the brain. These diets corrupt membrane amyloid beta homeostasis and determine neuron senescence and the aging process. Amyloid beta oligomers generated by cell membrane cholesterol and phospholipids interact with acute phase reactants that determine the benign or toxic amyloid beta conformational states. In yeast amyloid beta oligomers have different toxicities and are relevant to human amyloid beta oligomers in the brain. In mammalian cells the dynamic nature of the amyloid beta oligomer states may be altered by bacterial lipopolysaccharides that involve membrane amphiphilic and charge polarization. Lipopolysaccarides partition in cell membranes and its interaction with apolipoprotein E corrupts the peripheral amyloid beta metabolism with effects on toxic amyloid beta generation in the brain with relevance to neurodegeneration and Alzheimer’s disease. The role of atherogenic diets involve dysregulation of peripheral lipopolysaccharide metabolism with effects on apolipoprotein E/amyloid beta and albumin/amyloid beta interactions associated with increased lipopolysaccharides in brain cells that determine neuroinflammation with relevance to toxic amyloid beta behaviour and memory disorders.
Keywords: Diet; Amyloid beta; Lipopolysaccharides; Cholesterol; Neurodegeneration; NAFLD
Abbreviations
HDL: High Density Lipoprotein; LDL: Low Density Lipoprotein; LPS: Lipopolysaccharide; NAFLD: Non Alcoholic Fatty Liver Disease; AD: Alzheimer’s Disease; apo E: Apolipoprotein E; Aβ: Amyloid beta; APP: Acute Phase Proteins; PLTP: Phospholipid Transfer Protein; LBP: LPS Binding Protein; ABCA1: ATP Binding Cassette Transporter 1; EGCG: (-)-epigallactocatechin-3-gallate; LRP-1: Low Density Lipoprotein Receptor Related Protein 1; Sirt 1: Sirtuin 1; LDLr: Low Density Lipoprotein Receptor; BBB: Blood Brain Barrier; CD14: Cluster Of Differentiation 14; TLR-4: Toll-like Receptor 4
Introduction
Healthy diets such as low fat and high fibre diets [1] that prevent metabolic diseases and neurodegeneration and have become of critical interest to the prevention of Alzheimer’s disease (AD) a neurodegenerative condition that involves disturbances in multiple higher brain functions that include cognition and memory. The main constituent of senile plaques associated with AD is amyloid beta (Aβ) [2] that is a proteolytic product of the larger amyloid precursor protein (APP). APP is cleaved by three proteases classified as α, β and γ secretases in neurons with formation of Aβ by a two step process that involves the β-site cleaving enzyme (BACE) and the γ secretases. Intracellular cholesterol levels determine the increased production of Aβ 40 and Aβ 42 species [3,4] from APP with early stages of cholesterol involved in the altered apolipoprotein E (apo E) and Aβ interaction with the acceleration of amyloidogenesis [5]. Most proteins fold into their native structure with few intermediate structures that become toxic to cells. The understanding of Aβ and protein folding has increased with the ability of the peptide to self associate and determine the benign or toxic Aβ states that promote brain Aβ aggregation [6]. The Aβ self association properties in mouse and man differ [5] and similarities between yeast and mammalian cells in toxic Aβ oligomer species have been shown [7,8]. The aggregation of Aβ involves the electrostatic nature of oligomeric amyloid assemblies that leads to Aβ plaque with extensive brain pathology. In man unhealthy diets have attracted interest and determine the benign or toxic amyloid beta oligomers that involve abnormal apoE cell membrane interactions with neuronal death.
Healthy diets that contain unsaturated fat, fruit and fish (omega-3) are associated with the reversal of non alcoholic liver disease (NAFLD) with the prevention of accelerated brain ageing [9-13]. High fibre diets that contain phytosterols are important to lower brain membrane cholesterol [1] and promote Aβ metabolism by the liver with the prevention of oligomeric Aβ species generation in the brain. In aging and neurodegeneration healthy diets that protect neurons early in life have become important with the regulation of neuronal cholesterol by phytosterols that reduce increased Aβ production and its ability to self-associate with Aβ aggregation [1]. Low carbohydrate diets and diets without xenobiotics [14] improve the rapid transport of Aβ from the brain to the liver with the prevention of early neurodegeneration. Healthy diets further improve drug therapy (statins) by the up regulation of the low density lipoprotein (LDL) receptor that lower brain cholesterol and oligomers with effects on the reverse transport of Aβ across the blood brain barrier to the periphery (peripheral sink abeta hypothesis).
Unhealthy diets that include high protein intake, high fat and high sugar diets have been associated with circadian imbalances and AD [11,15]. Low fat diets improve the circadian rhythm and also lower the absorption of lipophilic xenobiotics that may enter the CNS and promote circadian disturbances and neurodegeneration [14]. Interest in low calorie diets have increased in the developing and developed world with activation of the calorie sensitive anti-aging gene Sirtuin 1 (Sirt 1), a nicotinamide dependent protein deacetylase that is involved in brain neuron proliferation [16], circadian rhythm and Aβ metabolism [11]. Unhealthy diets that contain excess fatty acids (palmitic acid) and glucose down regulate Sirt 1 with the development of NAFLD and AD. The role of nutrigenomics and metabolic health have become central to the treatment of AD with nutritional therapy involved in the activation of genes such as Sirt 1[12] involved in brain neuron cholesterol and the early stages of amyloidogenesis. Reduction in food intake and increased consumption of Sirt 1 activators such as leucine and pyruvic acid reverse the effects of unhealthy diets associated with toxic Aβ states and Aβ aggregation [11].
Neuroinflammation has now become closely linked to AD with unhealthy diets associated with an increase in acute phase reactants and cytokines in the blood plasma [5,17]. The links between acute phase proteins (APP) that prevent toxic Aβ generation are now linked to abnormalities in various cholesterol containing lipoproteins such as LDL and high density lipoproteins (HDL). Interests in food restriction and fasting that leads to the reduced transport of fat from the intestine to the plasma and liver has increased with relevance to reducing the detrimental effects of bacterial lipopolysacchardies (LPS) that are endotoxins released from the outer layer of gram negative bacteria in the gut [17,18] that are responsible for cholesterol dyshomeostasis and inflammatory acute phase reactants associated with α-synuclein and Aβ aggregation [17,18]. Diets and foods (high fat, dairy, meat) that contain gram negative bacteria produce LPS have now become of critical importance to organ disease in global communities [19] with connections between nutrient metabolism, dyslipidemia and amyloidosis. Diets that contain high fat, protein and carbohydrates delay the clearance of plasma LPS and the Aβ peptide with the possible induction of NAFLD and AD [20,21]. NAFLD in obese and diabetic individuals delays LPS clearance that provokes a strong inflammatory immune response with risk for endotoxemia with altered apo E regulated amyloidosis. Therefore the amount and nature of food eaten is connected to plasma LPS levels with relevance to the accelerated aging process that is linked to defective HDL cholesterol metabolism and toxic Aβ oligomer generation involved with memory disorders and brain amyloidosis.
LPS effects on apo E and albumin levels are associated with amyloid beta oligomer metabolism
The understanding of the role of the peripheral sink Aβ hypothesis in AD implicates LPS of central importance in the determination of lipoprotein metabolism, phospholipid transfer protein (PLTP) activity that involve lipoprotein/membrane cholesterol efflux and the role of various APP involved in Aβ aggregation (Figure 1). LPS are endotoxins and essential components of the outer membrane of gram negative bacteria and consist of covalently linked segments, surface carbohydrate polymer, core oligosaccharide and acylated glycolipid that can bind to cell membranes to alter membrane interactions [17]. Experiments in yeast that involve endocytic Aβ trafficking with toxic Aβ oligomer species generation [7,8,22] are different to Aβ metabolism in mammalian cells with the recent involvement of bacterial LPS that regulates mammalian cell membrane cholesterol and Aβ oligomer metabolism [17]. In obese and diabetic individuals the increased LPS levels are involved in the neutralization of apo E mediated hepatic clearance of abeta [5,17,19].
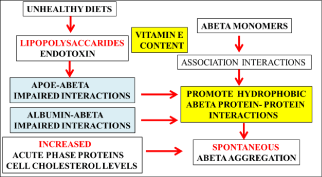
Figure 1: The effect of bacterial LPS on peripheral amyloid beta metabolism
involves abnormal cell membrane amyloid beta interactions with the promotion
of non-brownian amyloid beta-amyloid beta dynamics. 1. Toxic amyloid beta
oligomer species generation in mammalian cells is stimulated by bacterial
LPS that neutralizes the apo E mediated clearance and albumin mediated
clearance of peripheral abeta. 2. LPS increases acute phase proteins (APP)
in plasma with the abnormal regulation of peripheral abeta metabolism. 3.
LPS effects on membrane cholesterol efflux may involve corruption of apo
E-ABCA1 mediated cholesterol efflux with effects on cholesterol mediated
amyloidosis. 4. LPS and PLTP are involved in vitamin E, phospholipid and
amyloid beta transport in cell membranes with PLTP mediated transport of
bacterial LPS that determines membrane vitamin E content and amyloid beta
hydrophobic self association.
LPS regulate interactions between APP and Aβ oligomers and APP include gelsolin, serum amyloid protein A, serum amyloid protein, C-reactive protein, clusterin and transthyretin [17]. The plasma also include APP such as transferrin [23], albumin [23], phospholipid transfer protein (PLTP) [24,25] and LPS binding protein (LBP) [26] with albumin and transferrin closely associated with the peripheral Aβ metabolism [11,12,27-29]. Furthermore LPS and inflammation have shown to reduce the release of albumin [30,31] from the liver with effects on albumin mediated fatty acid transport [32] with plasma albumin (Figure 1) important to brain Aβ aggregation [28- 30]. In contrast to suppression of hepatic albumin by LPS the APP are involved with binding and inhibition of LPS mediated inflammatory processes [33].
LPS effects on ATP binding cassette transporter 1 (ABCA1) membrane cholesterol efflux may involve corruption of apo E-ABCA1 mediated cholesterol efflux [34,35] with effects on cholesterol mediated amyloidosis (Figure 1). The role of apo E is intimately involved in ABCA1 mediated cholesterol efflux by activation of PLTP activity [10]. PLTP is central to hepatic ABCA1 mediated cholesterol efflux [36,37] to HDL with apolipoprotein B lipoprotein secretion [38] associated with LPS transport. PLTP is involved in vitamin E, phospholipid and Aβ transport in cell membranes [39- 44] with PLTP mediated transport of bacterial LPS [45-50] that partitions in membranes and determines membrane vitamin E content and monomeric Aβ hydrophobic self association (Figure 1) with toxic Aβ generation [23,51]. In obese and diabetic plasma the elevated LPS levels may closely be involved with APP regulation and toxic Aβ intermediates (non-brownian dynamics) associated with dyslipidemia and NAFLD in these individuals [17,19,20]. Acute phase reactants such as PLTP (Mwt 78 kda) and LBP (Mwt 65 kda) belong to a common gene family for lipid binding proteins with both proteins associated with albumin in the LPS transport between lipoproteins and hepatic cell membranes [52-54]. Increased levels of vitamin E administration in rats prevent LPS mediated hepatic damage [55] with facilitation of peripheral abeta clearance.
Alpha-synuclein binding to cholesterol membranes in mammalian cells regulates membrane cholesterol homeostasis and is linked to LPS and delayed Aβ oligomer metabolism [18]. In yeast alpha-synuclein prevents toxic abeta species generation [8] with the yeast alpha synuclein and Aβ interaction independent from inflammatory processes from LPS on Aβ oligomer generation as in mammalian cells [17]. In yeast apart from ergosterol [22] and mammalian cells (cholesterol) the alpha synuclein modification of Aβ oligomer is also regulated by other membrane lipids [18] with relevance to alpha synuclein and lipid involvement in benign and toxic oligomer Aβ dynamics in mammalian cells.
LPS induces inflammation and dyslipidemia with increased risk for NAFLD and amyloidosis
In AD the low HDL are associated with the increased risk for neurodegeneration and amyloidosis. Increased LPS and elevated PLTP levels in obese and diabetic individuals may be responsible for inflammation and the dyslipidemia [10] with low HDL, high LDL levels and increased triglyceride levels relevant to increased risk for AD [10]. Furthermore lipoproteins have been shown to be essential for the receptor mediated endocytosis of LPS in both the liver, macrophages and artery wall [17,19,56]. Connections between dyslipidemia and poor LPS clearance (Figure 2) are related to the induction of NAFLD by LPS and relevant to increased intestinal transport of LPS to the plasma and liver [20]. Increased dietary fat and chylomicron production has been closely connected to increased plasma LPS levels [56] with the development of NAFLD [11] and cardiovascular disease [57]. Interests in the role of LPS induction of chronic diseases may be related to LPS mediated mitochondrial apoptosis with decreased fatty acid metabolism [58,59]. The links between defects in cellular lipid metabolism and promotion of Aβ oligomer species indicate LPS to be involved in both dyslipidemia and amyloidosis (Figure 2). The increased LPS plasma levels and its hepatic lipoprotein mediated transport into endosome and lysosomes are critical to reduction of plasma LPS levels. The high fat and high cholesterol diets that stimulate LPS absorption are involved with the decreased hepatic intracellular lipid/LPS metabolism and linked to hepatic inflammation and the induction of NAFLD.
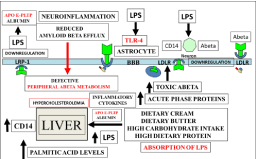
Figure 2: Increased dietary fat and chylomicron production are connected
to increased plasma LPS levels with the development of NAFLD and AD. 1.
Diets that are high in fat reduce the plasma clearance of LPS by the liver. 2.
The links between defects in peripheral lipid metabolism and promotion of
amyloid beta aggregation indicate LPS to be involved in both dyslipidemia
and corruption of the transport of amyloid beta from the brain to the blood
plasma. 3. LPS mediated neuroinflammation of astrocytes is involved in
the defective clearance of amyloid beta from neurons. 4. Downregulation of
receptors such as the LDLr and LRP-1 are associated with delayed LPS and
amyloid beta transport connected to low HDL and high LDL cholesterol levels.
The nuclear receptor Sirt 1 is associated with fatty acid metabolism, mitochondrial biogenesis, insulin resistance, NAFLD and amyloidosis and is clearly corrupted by LPS with acceleration in the various chronic diseases and neurodegeneration [12]. Interest in LPS regulation of nuclear Sirt 1 has attracted interest with its involvement in alpha synuclein and Aβ metabolism [18]. In contrast to the studies in yeast with (-)-epigallactocatechin-3-gallate (EGCG) and its arrest of oligomeric Aβ species the effect of the EGCG in mammalian cells have detrimental effects on the post-transcriptional regulation of the p53 tumour suppressor protein associated with the downregulation Sirt 1 expression and reduced hepatic Sirt 1 deacetylase activity connected to defective alpha-synuclein and Aβ metabolism [12,18]. LPS has also been associated with zinc deficiency [60-62] and connected to apo E function [63] Aβ dynamics with aggregation [64,65]. Nutritional intake of zinc has become important to global populations with zinc essential to maintain cellular Sirt 1 activity [66], albumin [67], vitamin E [68,69] and HDL levels [70,71].
In aging and AD membrane changes in LPS related toxicity may influence neuron membrane cholesterol by binding to cell membranes with altered membrane interactions that possibly involve the role of LPS in Aβ aggregation and fibril formation. LPS related effects on neuron membrane cholesterol may also involve its neutralization of apo E that is closely linked to brain cholesterol homeostasis and the reverse brain Aβ transport to the periphery (peripheral sink abeta hypothesis). LPS related neurodegeneration may involve specifically reverse cholesterol transport with the disruption of LXR-ABCA1 interactions that determine cell cholesterol dysregulation involved in the generation of toxic Aβ species that occur in the early stages of AD. In the brain the CD14 receptor is referred to as the LPS receptor (Figure 2) and involved with Aβ metabolism [17]. The cluster of differentiation 14 (CD14) receptor assists in the co-ordination of the microglia that promotes Aβ mediated and oxidative neuron death [72].
Research in the role of LPS in the peripheral sink Aβ hypothesis has escalated with the improved understanding of the LPS effects on astrocytes and neurons in the brain and effects on the transport across the blood brain barrier (BBB). Studies have indicated that LPS was associated with impaired Aβ efflux across the BBB with the downregulation of the low density lipoprotein receptor related protein 1 (LRP-1) that has previously been shown to be critical to the Aβ efflux (Figure 2) from the brain [73]. The effects of systemic LPS on neuroinflammation [74,75] and disturbed Aβ homeostasis are similar to the disease progression in AD with the effects of LPS on cognitive impairment relevant to Aβ generation [76,77]. Further experiments with LPS implicate its role in memory processing deficits and neuronal death [78-80]. Toll-like receptor 4(TLR-4) that is closely linked to neuroinflammation and AD is also activated by LPS in mouse and human astrocytes [81,82]. Activation of inflammation in astrocytes by LPS (Figure 2) corrupts the important role of astrocytes in the metabolism of neuronal Aβ [10] with the development of AD. As LPS concentrations increase in plasma the unbound LPS can spontaneously insert in cell lipid bilayers with increased cell membrane LPS content that involve the cholesterol/sphingomyelin domains [17,19] with the hydrophilic polysaccharide chain exposed to the plasma. Neutralization of apo E and Aβ cell membrane interactions by LPS may not only involve the presence of excess membrane LPS but also low plasma Zn2+ levels [60-62] that may lead to abnormal apo E and Aβ cell membrane interactions [5,17,19]. LPS binds to LBP and interactions with the CD14 receptor in macrophage membranes stimulates the release of pro-inflammatory cytokines such as tumour necrosis factor, interleukin 1 and interleukin 6. In the liver cytokines such as interleukin 6 stimulate the release of CD14 (acute phase protein) into the circulation [83]. The binding of LPS to various lipoprotein membranes modulates the acute phase reactions in the periphery and delays LPS binding to the CD14 receptor. In apo E knockout mice susceptibility to endotoxemia implicate the role of apo E lipoproteins in LPS transport and in the LDL receptor knockout mice the increased LDL cholesterol concentrations (Figure 2) delay the LPS mediated inflammatory response [84,85]. LDL receptor deficiency has been associated with astrocytosis with increased amyloid deposition that implicate LPS in the LDLr regulation of astrocyte-neuron Aβ metabolism [10,86]. Saturated fatty acids such as palmitic and myristic acid raise LDL cholesterol levels [87-89] with increased palmitic acid levels (butter, cream, high carbohydrate intake) sensitive to the downregulation of the LDL receptor (Figure 2) with relevance to the peripheral clearance of toxic abeta and the promotion of neurodegeneration and brain amyloidosis [10]. The low HDL associated with the increased risk for AD and its close connections to disturbed cholesterol metabolism possibly determine the benign or toxic Aβ conformational states. The Western diet that is high in fat determine the different toxicities to human Aβ oligomers with the consumption of phosphatidylinositol [19] important in the maintenance of plasma HDL levels and the prevention of toxic Aβ generation with reversal of abeta aggregation [5]. Furthermore therapeutic phytosterol intake (approx. 2 gm/day) is important to diabetes treatment and prevention of organ dysfunction [19] with phytosterol intake closely linked to the maintenance of hepatic cholesterol and Aβ metabolism in metabolic disease via ABCA1 pathways [1]. Elevated plasma LPS levels corrupt phytosterol-ABCA1 [1] and PLTP pathways [90] that are critical to formation of HDL, toxic Aβ oligomer species and interactions with APP in protein misfolding [5,17] may play an early role in the development of neurodegenerative disease. Furthermore the direct therapeutic effects of phytosterol on Aβ reduction in the brain [91] may be affected by increased brain LPS levels that induce astrocyte inflammation related to defective neuronal Aβ metabolism and elevated phytosterol intake as persistent nutritional therapy may be harmful with increased brain phytosterol accumulation associated with population ageing (> 85 years)and AD [92,93].
Conclusion
Interest in metabolic and neurodegenerative diseases have increased in global communities and implicate high fat/cholesterol diets and alcohol to be responsible for increased plasma LPS levels that are involved in the induction of NAFLD and AD. The links between lipoprotein metabolism and abnormal apo E-PLTP interactions in AD clearly indicate that dyslipidemia is associated with delayed hepatic LPS clearance with toxic Aβ generation. Bacterial LPS corruption of the astrocyte-neuron interaction by inflammatory processes delays brain Aβ clearance with increased brain amyloid plaque development in various communities associated with excessive feeding and abnormal liver lipid metabolism. Reversal of NAFLD by healthy diets such as low fat (palmitic acid) and high fibre diets that contain appropriate phytosterol, vitamin E and phosphatidylinositol (2 gm/ day) increase LPS and Aβ metabolism and reduce transport of LPS to the brain with improved memory and cognition connected to therapeutic plasma albumin levels.
Acknowledgement
This work was supported by grants from Edith Cowan University, the McCusker Alzheimer’s Research Foundation and the National Health and Medical Research Council.
References
- Martins IJ, Fernando W. High Fibre Diets and Alzheimer’s Disease. Food and Nutrition Sciences. 2014; 5: 410-424.
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985; 82: 4245-4249.
- Fantini J, Yahi N, Garmy N. Cholesterol accelerates the binding of Alzheimer's β-amyloid peptide to ganglioside GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front Physiol. 2013; 4: 1.
- Amaro M, Kubiak-Ossowska K, Birch DJS and Rolinski OL. Initial stages of beta-amyloid Aβ1-40 and Aβ1-42 oligomerization observed using fluorescence decay and molecular dynamics analyses of tyrosine. Methods Appl Fluoresc. 2013; 1: 015006.
- Martins IJ, Gupta V, Wilson AC, Fuller SJ, Martins RN. Interactions between Apo E and Amyloid Beta and their Relationship to Nutriproteomics and Neurodegeneration. Current Proteomics. 2014; 11: 173-183.
- Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008; 283: 29639-29643.
- Krishnan R, Goodman JL, Mukhopadhyay S, Pacheco CD, Lemke EA, Deniz AA, et al. Conserved features of intermediates in amyloid assembly determine their benign or toxic states. Proc Natl Acad Sci U S A. 2012; 109: 11172-11177.
- Treusch S, Hamamichi S, Goodman JL, Matlack KE, Chung CY, Baru V, et al. Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science. 2011; 334: 1241-1245.
- Martins IJ. The Global Obesity Epidemic is related to Stroke, Dementia and Alzheimer’s disease. JSM Alzheimer’s Dis Related Dementia. 2014; 1: 1010.
- Martins IJ, Creegan R. Links between Insulin Resistance, Lipoprotein Metabolism and Amyloidosis in Alzheimer’s Disease. Health. 2014; 6:1549-1579.
- Martins, IJ. Induction of NAFLD with Increased Risk of Obesity and Chronic Diseases in Developed Countries. OJEMD. 2014; 4: 90-110.
- Martins IJ. Unhealthy Nutrigenomic Diets Accelerate NAFLD and Adiposity in Global communities. J Mol Genet Med. 2015; 9: 162.
- Martins IJ, Wood KM, Fernandis AZ, Taddei K, Martins RN. Anti-Oxidative Acyl CoA Cholesterol Acyltransferase Inhibitor AVASIMIBE Reduces the Impact of a High Cholesterol Diet on Brain Lipid Peroxidation in Mice. ADPD. 2013. Florence. www.kenes.com.
- Martins IJ. Increased Risk for Obesity and Diabetes with Neurodegeneration in Developing Countries. J Mol Genet Med. 2013; S1: 001.
- Hastings MH, Reddy AB, Maywood ES. A clockwork web: circadian timing in brain and periphery, in health and disease. Nat Rev Neurosci. 2003; 4: 649-661.
- Herskovits AZ, Guarente L. Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res. 2013; 23: 746-758.
- Martins IJ. LPS regulates apolipoprotein E and amyloid beta interactions with effects on acute phase proteins and amyloidosis. AAR. 2015; 4: 69-77.
- Martins IJ. Diabetes and cholesterol dyshomeostasis involves abnormal a-synuclein and amyloid beta transport in neurodegenerative diseases. Austin Alzheimer's and Parkinson's Disease. 2015. [In Press].
- Martins IJ. Diabetes and Organ dysfunction in the developing and developed world. Global Journal of Medical Research. 2015; 15: 1-8.
- Ilan Y. Leaky gut and the liver: a role for bacterial translocation in nonalcoholic steatohepatitis. World J Gastroenterol. 2012; 18: 2609-2618.
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007; 56: 1761-1772.
- Souza CM, Schwabe TM, Pichler H, Ploier B, Leitner E, Guan XL, et al. A stable yeast strain efficiently producing cholesterol instead of ergosterol is functional for tryptophan uptake, but not weak organic acid resistance. Metab Eng. 2011; 13: 555-569.
- Ramamoorthy A, Lim MH. Structural characterization and inhibition of toxic amyloid-β oligomeric intermediates. Biophys J. 2013; 105: 287-288.
- Pussinen PJ, Malle E, Metso J, Sattler W, Raynes JG, Jauhiainen M. Acute-phase HDL in phospholipid transfer protein (PLTP)-mediated HDL conversion. Atherosclerosis. 2001; 155: 297-305.
- Pussinen PJ, Metso J, Malle E, Barlage S, Palosuo T, Sattler W, et al. The role of plasma phospholipid transfer protein (PLTP) in HDL remodeling in acute-phase patients. Biochim Biophys Acta. 2001; 1533: 153-163.
- Tobias PS, Soldau K, Ulevitch RJ. Isolation of a lipopolysaccharide-binding acute phase reactant from rabbit serum. J Exp Med. 1986; 164: 777-793.
- Milojevic J, Melacini G. Stoichiometry and affinity of the human serum albumin-Alzheimer's Aβ peptide interactions. Biophys J. 2011; 100: 183-192.
- Costa M, Ortiz AM, Jorquera JI. Therapeutic albumin binding to remove amyloid-β. J Alzheimers Dis. 2012; 29: 159-170.
- Ruot B, Breuillé D, Rambourdin F, Bayle G, Capitan P, Obled C. Synthesis rate of plasma albumin is a good indicator of liver albumin synthesis in sepsis. Am J Physiol Endocrinol Metab. 2000; 279: E244-251.
- Wang X, Li W, Lu J, Li N, Li J. Lipopolysaccharide suppresses albumin expression by activating NF-kappaB in rat hepatocytes. J Surg Res. 2004; 122: 274-279.
- Don BR, Kaysen G. Serum albumin: relationship to inflammation and nutrition. Semin Dial. 2004; 17: 432-437.
- van der Vusse GJ. Albumin as fatty acid transporter. Drug Metab Pharmacokinet. 2009; 24: 300-307.
- Triantafilou M, Mouratis MA, Lepper PM, Haston RM, Baldwin F, Lowes S, et al. Serum proteins modulate lipopolysaccharide and lipoteichoic acid-induced activation and contribute to the clinical outcome of sepsis. Virulence. 2012; 3: 136-145.
- Krimbou L, Denis M, Haidar B, Carrier M, Marcil M, Genest J Jr. Molecular interactions between apoE and ABCA1: impact on apoE lipidation. J Lipid Res. 2004; 45: 839-848.
- Zhao Y, Chen X, Yang H, Zhou L, Okoro EU, Guo Z. A novel function of apolipoprotein E: upregulation of ATP-binding cassette transporter A1 expression. PLoS One. 2011; 6: e21453.
- Oram JF, Wolfbauer G, Tang C, Davidson WS, Albers JJ. An amphipathic helical region of the N-terminal barrel of phospholipid transfer protein is critical for ABCA1-dependent cholesterol efflux. J Biol Chem. 2008; 283: 11541-11549.
- Oram JF, Wolfbauer G, Vaughan AM, Tang C, Albers JJ. Phospholipid transfer protein interacts with and stabilizes ATP-binding cassette transporter A1 and enhances cholesterol efflux from cells. J Biol Chem. 2003; 278: 52379-52385.
- Lagrost L. Plasma phospholipid transfer protein: a multifaceted protein with a key role in the assembly and secretion of apolipoprotein B-containing lipoproteins by the liver. Hepatology. 2012; 56: 415-418.
- Desrumaux C, Risold PY, Schroeder H, Deckert V, Masson D, Athias A, et al. Phospholipid transfer protein (PLTP) deficiency reduces brain vitamin E content and increases anxiety in mice. FASEB J. 2005; 19: 296-297.
- Martins IJ, Hopkins L, Joll CA, Redgrave TG. Interactions between model triacylglycerol-rich lipoproteins and high-density lipoproteins in rat, rabbit and man. Biochim Biophys Acta. 1991; 1081: 328-338.
- Martins IJ, Redgrave TG. The effect of insulin deficiency on the plasma clearance and exchange of high-density-lipoprotein phosphatidylcholine in rats. Biochem J. 1992; 281: 851-857.
- Nishida Y, Ito S, Ohtsuki S, Yamamoto N, Takahashi T, Iwata N, et al. Depletion of vitamin E increases amyloid beta accumulation by decreasing its clearances from brain and blood in a mouse model of Alzheimer disease. J Biol Chem. 2009; 284: 33400-33408.
- Desrumaux C, Pisoni A, Meunier J, Deckert V, Athias A, Perrier V, et al. Increased amyloid-β peptide-induced memory deficits in phospholipid transfer protein (PLTP) gene knockout mice. Neuropsychopharmacology. 2013; 38: 817-825.
- Muñoz FJ, Solé M, Coma M. The protective role of vitamin E in vascular amyloid beta-mediated damage. Subcell Biochem. 2005; 38: 147-165.
- Hailman E, Albers JJ, Wolfbauer G, Tu AY, Wright SD. Neutralization and transfer of lipopolysaccharide by phospholipid transfer protein. J Biol Chem. 1996; 271: 12172-12178.
- Levels JH, Marquart JA, Abraham PR, van den Ende AE, Molhuizen HO, van Deventer SJ, et al. Lipopolysaccharide is transferred from high-density to low-density lipoproteins by lipopolysaccharide-binding protein and phospholipid transfer protein. Infect Immun. 2005; 73: 2321-2326.
- Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008; 5: 37.
- Bitting L, Naidu A, Cordell B, Murphy GM Jr. Beta-amyloid peptide secretion by a microglial cell line is induced by beta-amyloid-(25-35) and lipopolysaccharide. J Biol Chem. 1996; 271: 16084-16089.
- Navab M, Hough GP, Van Lenten BJ, Berliner JA, Fogelman AM. Low density lipoproteins transfer bacterial lipopolysaccharides across endothelial monolayers in a biologically active form. JClin Invest. 1988; 81: 601–605.
- Van Lenten BJ, Fogelman AM, Haberland ME, Edwards PA. The role of lipoproteins and receptor-mediated endocytosis in the transport of bacterial lipopolysaccharide. Proc Natl Acad Sci U S A. 1986; 83: 2704-2708.
- Santucci R, Sinibaldi F, Fiorucci L. Protein folding, unfolding and misfolding: role played by intermediate States. Mini Rev Med Chem. 2008; 8: 57-62.
- Kirschning CJ, Au-Young J, Lamping N, Reuter D, Pfeil D, Seilhamer JJ, et al. Similar organization of the lipopolysaccharide-binding protein (LBP) and phospholipid transfer protein (PLTP) genes suggests a common gene family of lipid-binding proteins. Genomics. 1997; 46: 416-425.
- Bingle CD, Craven CJ. Meet the relatives: a family of BPI- and LBP-related proteins. Trends Immunol. 2004; 25: 53-55.
- Gioannini TL, Zhang D, Teghanemt A, Weiss JP. An essential role for albumin in the interaction of endotoxin with lipopolysaccharide-binding protein and sCD14 and resultant cell activation. J Biol Chem. 2002; 277: 47818-47825.
- Bharrhan S, Chopra K, Rishi P. Vitamin E Supplementation Modulates Endotoxin-induced Liver Damage in a Rat Model Am. J Biomed Sci. 2010; 2: 51-62.
- Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009; 50: 90-97.
- Charalambous BM, Stephens RC, Feavers IM, Montgomery HE. Role of bacterial endotoxin in chronic heart failure: the gut of the matter. Shock. 2007; 28: 15-23.
- Noh H, Jeon J, Seo H. Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem Int. 2014; 69: 35-40.
- Kuwabara T, Imajoh-Ohmi S. LPS-induced apoptosis is dependent upon mitochondrial dysfunction. Apoptosis. 2004; 9: 467-474.
- Pitt JA, Zoellner MJ, Carney EW. In vivo and in vitro developmental toxicity in LPS-induced zinc-deficient rabbits. Reprod Toxicol. 1997; 11: 771-779.
- Shea-Budgell M, Dojka M, Nimmo M, Lee D, Xu Z. Marginal zinc deficiency increased the susceptibility to acute lipopolysaccharide-induced liver injury in rats. Exp Biol Med (Maywood). 2006; 231: 553-558.
- Wang J, Jiao L, Ma J, Wu C, Wang K, Wang M. Effects of intravenous infusion of lipopolysaccharide on plasma micromineral, magnesium, and cytokine concentrations and serum cortisol concentrations in lactating goats. Am J Vet Res. 2007; 68: 529-534.
- Xu H, Finkelstein DI, Adlard PA. Interactions of metals and Apolipoprotein E in Alzheimer's disease. Front Aging Neurosci. 2014; 6: 121.
- Rezaei-Ghaleh N, Giller K, Becker S, Zweckstetter M. Effect of zinc binding on β-amyloid structure and dynamics: implications for Aβ aggregation. Biophys J. 2011; 101: 1202-1211.
- Miller Y, Ma B, Nussinov R. Zinc ions promote Alzheimer Abeta aggregation via population shift of polymorphic states. Proc Natl Acad Sci U S A. 2010; 107: 9490-9495.
- Zee RS, Pimentel DR, Hou XL, Zang M, Yao CX, O'Connor PB, et al. Sirtuin-1 zinc thiolatecenter is a direct molecular target of oxidants. In: Society for Free Radical Biology and Medicine's 15th Annual Meeting, Indianapolis, IN, USA. Published in: Free Radical Biology and Medicine. 2008; 45 (Supplement). S76.
- McMillan EM, Rowe DJ. Plasma zinc-serum albumin correlation: relevance to assessment of zinc status in humans. Clin Exp Dermatol. 1982; 7: 599-604.
- Bunk MJ, Dnistrian AM, Schwartz MK, Rivlin RS. Dietary zinc deficiency decreases plasma concentrations of vitamin E. Proc Soc Exp Biol Med. 1989; 190: 379-384.
- Goode HF, Purkins L, Heatley RV, Kelleher J. The effect of dietary vitamin E deficiency on plasma zinc and copper concentrations. Clin Nutr. 1991; 10: 233-235.
- Goodwin JS, Hunt WC, Hooper P, Garry PJ. Relationship between zinc intake, physical activity, and blood levels of high-density lipoprotein cholesterol in a healthy elderly population. Metabolism. 1985; 34: 519-523.
- Yasutake K, Kohjima M, Nakashima M, Kotoh K, Nakamuta M, Enjoji M. Nutrition therapy for liver diseases based on the status of nutritional intake. Gastroenterol Res Pract. 2012; 2012: 859697.
- Bate C, Veerhuis R, Eikelenboom P, Williams A. Microglia kill amyloid-beta1-42 damaged neurons by a CD14-dependent process. Neuroreport. 2004; 15: 1427-1430.
- Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA. Lipopolysaccharide impairs amyloid β efflux from brain: altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood-brain barrier. J Neuroinflammation. 2012; 9: 150.
- Murray CL, Skelly DT, Cunningham C. Exacerbation of CNS inflammation and neurodegeneration by systemic LPS treatment is independent of circulating IL-1β and IL-6. J Neuroinflammation. 2011; 8: 50.
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007; 55: 453-462.
- Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008; 5: 37.
- Llewellyn DJ, Langa KM, Friedland RP, Lang IA. Serum albumin concentration and cognitive impairment. Curr Alzheimer Res. 2010; 7: 91-96.
- Vasconcelos AR, Yshii LM, Viel TA, Buck HS, Mattson MP, Scavone C, et al. Intermittent fasting attenuates lipopolysaccharide-induced neuroinflammation and memory impairment. J Neuroinflammation. 2014; 11: 85.
- Tanaka S, Ide M, Shibutani T, Ohtaki H, Numazawa S, Shioda S, et al. Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J Neurosci Res. 2006; 83: 557-566.
- Sell KM, Crowe SF, Kent S. Lipopolysaccharide induces memory-processing deficits in day-old chicks. Pharmacol Biochem Behav. 2001; 68: 497-502.
- Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol Biochem. 2007; 20: 947-956.
- Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011; 59: 242-255.
- Bas S, Gauthier BR, Spenato U, Stingelin S, Gabay C. CD14 is an acute-phase protein. J Immunol. 2004; 172: 4470-4479.
- de Bont N, Netea MG, Demacker PN, Verschueren I, Kullberg BJ, van Dijk KW, et al. Apolipoprotein E knock-out mice are highly susceptible to endotoxemia and Klebsiella pneumoniae infection. J Lipid Res. 1999; 40: 680-685.
- Netea MG, Demacker PN, Kullberg BJ, Boerman OC, Verschueren I, Stalenhoef AF, et al. Low-density lipoprotein receptor-deficient mice are protected against lethal endotoxemia and severe gram-negative infections. J Clin Invest. 1996; 97: 1366-1372.
- Katsouri L, Georgopoulos S. Lack of LDL receptor enhances amyloid deposition and decreases glial response in an Alzheimer's disease mouse model. PLoS One. 2011; 6: e21880.
- Zock PL, de Vries JH, Katan MB. Impact of myristic acid versus palmitic acid on serum lipid and lipoprotein levels in healthy women and men. Arterioscler Thromb. 1994; 14: 567-575.
- Mustad VA, Ellsworth JL, Cooper AD, Kris-Etherton PM, Etherton TD. Dietary linoleic acid increases and palmitic acid decreases hepatic LDL receptor protein and mRNA abundance in young pigs. J Lipid Res. 1996; 37: 2310-2323.
- Chavez-Tapia NC, Rosso N, Tiribelli C. Effect of intracellular lipid accumulation in a new model of non-alcoholic fatty liver disease. BMC Gastroenterol. 2012; 12: 20.
- Yamashita S, Sakai N, Hirano K, Ishigami M, Maruyama T, Nakajima N, et al. Roles of plasma lipid transfer proteins in reverse cholesterol transport. Front Biosci. 2001; 6: D366-387.
- Burg VK, Grimm HS, Rothhaar TL, Grösgen S, Hundsdörfer B, Haupenthal VJ, et al. Plant sterols the better cholesterol in Alzheimer's disease? A mechanistical study. J Neurosci. 2013; 33: 16072-16087.
- McDaniel AL, Alger HM, Sawyer JK, Kelley KL, Kock ND, Brown JM, et al. Phytosterol feeding causes toxicity in ABCG5/G8 knockout mice. Am J Pathol. 2013; 182: 1131-1138.
- Vanmierlo T, Rutten K, van Vark-van der Zee LC, Friedrichs S, Bloks VW, Blokland A, et al. Cerebral accumulation of dietary derivable plant sterols does not interfere with memory and anxiety related behavior in Abcg5-/- mice. Plant Foods Hum Nutr. 2011; 66: 149-156.