
Review Article
Austin J Clin Neurol 2015; 2(10): 1082.
Peroxidation and Halogenation Stress: Windows to a Better Understanding of Sporadic Parkinson’s Disease
Fernández E*
Department of Medical Physiology and Biophysics, University of Seville, Spain
*Corresponding author: Emilio Fernandez, Department of Medical Physiology and Biophysics, Laboratory of Molecular Neurology and Neurophysiology (BIO127), School of Medicine, Universidad de Sevilla, Av. Sanchez Pizjuan 4, E-41009 Sevilla, Spain
Received: September 11, 2015; Accepted: November 30, 2015; Published: December 02, 2015
Abstract
Oxidative stress is considered as an important pathogenic mechanism in Parkinson’s disease. Two types of oxidative stress, peroxidation and halogenation stress, are gaining increasing importance as biochemical windows to a better understanding of the pathogenesis of this disease. Peroxidation stress is due to excess of hydrogen peroxide, and it is related to the presence of many peroxidation-related molecular markers in Parkinsonian patients, and to misfolding of proteins. Peroxidation-induced misfolded proteins show altered functionality, such as loss of neuroprotective activity and tendency to form proteinaceous aggregates inside neurons. Peroxidation stress is also detected by a loss of activity of the main hydrogen peroxide scavengers in cerebrospinal fluid of patients. Altered hydrogen peroxide scavenging also leads to halogenation stress. Halogenation stress is characterized by the excess of halogenated molecules such as hypohalous acids, haloamines and halogenated proteins. Halogenated amines and proteins are thought to be deleterious for neurons, and they could play an important role in the etiology of Parkinson’s disease.
Keywords: Oxidative stress; Peroxidation; Halogenation; Hydrogen peroxide; Haloamine; Halogenated protein; Parkinson’s Disease
Abbreviations
AOPP: Advanced Oxidation Protein Products; αSYN: α-Synuclein; CNS: Central Nervous System; CSF: Cerebrospinal Fluid; EOP: Eosinophil Peroxidase; GPx: Glutathione-Peroxidase; GR: Glutathione-Reductase; GSH: Glutathione; GSSG: Glutathione Disulfide; GST: Glutathione-S-Transferase; 8-OHdG: 8-Hydroxyguanosine; H2O2: Hydrogen Peroxide; HSA: Human Serum Albumin; LPO: Lactoperoxidase; MDA: Malondialdehyde; MPO: Myeloperoxidase; NADPH: Nicotinamide Adenine Dinucleotide Phosphate; NOX: Nicotinamide Adenine Dinucleotide Phosphate Oxidase; O2: Oxygen; •O2 −: Superoxide Anion; •OH: Hydroxyl Ion; PARK2: Parkin; PD: Parkinson’s Disease; PRDx: Peroxiredoxin; SULT: Sulfotransferase; SH: Thiol Group; SOD: Superoxide Dismutase; SOH: Sulfenic Acid; TPO: Thyroperoxidase
Introduction
Oxidative stress, which is defined as an imbalance between the production of reactive oxidative species and anti-oxidant mechanisms, is considered as an important pathogenic mechanism in Parkinson’s disease (PD) and other neurodegenerative diseases [1-4]. Two types of oxidative stress, peroxidation and halogenation stress, are gaining increasing importance as biochemical windows to a better understanding of the pathogenesis of PD.
Peroxidation stress, defined as excess of production of hydrogen peroxide (H2O2) or loss of normal H2O2 scavenging, is an important type of oxidative stress. Peroxidation stress is related to the overproduction of free radicals and reactive oxygen species (ROS), which are otherwise products of the normal cellular metabolism. Superoxide ions (•O2 −) are the most important ROS, and they are produced in the mitochondrial matrix or through the NADPHoxidase (NOX) enzymes, mostly located extracellularly and attached to the plasmatic membrane. The first line of defense of the organism is to minimize the production of •O2 − in the mitochondria, because metabolic O2 is converted directly into water thanks to the action of cytochromes of the electronic chain. However, around 0.1-1% of electrons transferred to O2 can yield •O2 −. Superoxide anions are then catalyzed into hydrogen peroxide through several superoxide dismutases (SODs): SOD1 or copper-Zinc SOD of the cytosol, SOD2 or manganese SOD of the mitochondria, and SOD3 or extracellular SOD. H2O2is then converted into water after reacting with reduced glutathione (GSH), under the control of glutathione-peroxidase (GPx). The H2O2 produced from •O2 − is also eliminated through the action of catalases and peroxiredoxins (PRDxs). Catalases carry out dismutation reactions, and most catalases are heme-containing enzymes. Peroxiredoxins are cysteine-dependent or thiol (SH~) peroxidases. Hydrogen peroxide oxidizes the cysteine of PRDxs to protein sulfenic acid (SOH~PRDx). To sum up, SODs scavenge superoxide anions, and the most important scavengers of hydrogen peroxide are GPx, catalases and PRDxs (Figure 1). It can be concluded that a failure in these anti-oxidant defenses could lead to oxidative stress, with overproduction of ROS, mainly •O2 − or H2O2.
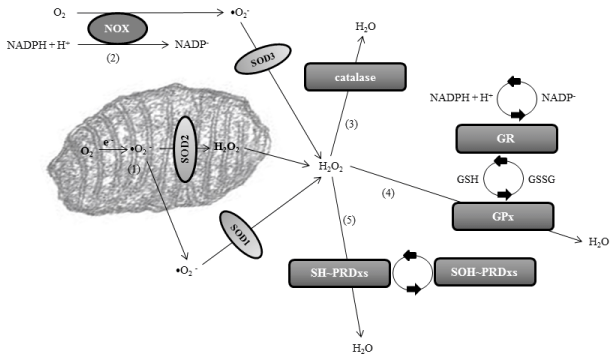
Figure 1: Pathways of normal redox homeostasis of superoxide anion and hydrogen peroxide leading to the final formation of water. The main production of
superoxide anion (•O2
−) is in the mitochondrial matrix [1] or through the NADPH-oxidase (NOX) enzymes [2]. Superoxide anions are catalyzed into hydrogen
peroxide through several superoxide dismutase’s (SODs): SOD1 or copper-Zinc SOD of the cytosol, SOD2 or manganese SOD of the mitochondria, and SOD3
or extracellular SOD. Hydrogen peroxide is then converted into water through catalases [3], glutathione-peroxidase (GPx) [4], and peroxiredoxins (PRDxs) [5].
Glutathione-peroxidase is integrated in the glutathione (GSH) system, where NADPH acts as the electron donor through glutathione-reductase (GR). GSH is
transformed in glutathione disulfide (GSSG) detoxifying the cells. Peroxirredoxins are thiol (SH~) peroxidases. Hydrogen peroxide oxidizes the cysteine of PRDxs
to protein sulfenic acid (SOH~). Abbreviation: NADPH, nicotinamide adenine dinucleotide phosphate.
Overproduction of hydrogen peroxide is also biochemically linked to halogenation stress, another type of oxidative stress that is characterized by excess of halogenated radicals or reduced scavenging of halogenated molecules. Halogenation reactions are mediated by haloperoxidases, a group of enzymes that mediate the conversion of hydrogen peroxide into hypohalous acids, such as hypochlorous or hypoiodous acids, after the incorporation of halogen elements such as chloride or iodide [5,6]. Hypohalous acids can react with primary amines to form halogenated amines (haloamines) such as chlorinated or iodinated amines. Normally hypohalous acids and haloamines are scavenged by glutathione-S-transferase, leading to the formation of oxyacids and sulfoxides. Haloamines can also be eliminated by sulfation through the action of sulfotransferases or SULTs [7], or after the formation of nitriles and aldehydes, although these latter reactions are slow in normal physiology. However if there is peroxidation stress and excess of H2O2, as it seems to be the case in PD [1-4], the enhanced formation of hypohalous acids and halogenated amines would lead to the formation of halogenated proteins, due to the incorporation of halogens at tyrosine and lysine residues of proteins. In addition, halogenated proteins could be subjected to degradation by proteasome, resulting in further release of free haloamines [8]. Haloamines in turn increase ROS production, thereby inducing a vicious cycle of oxidation and toxicity.
Peroxidation and Parkinson’s disease
Peroxidation stress is detected in brain tissue, cerebrospinal fluid (CSF) and blood, among other fluids, of PD patients. Analysis of substantia nigra tissue indicates that this nucleus is subjected to oxidative stress because it shows enhancement of several peroxidative markers such as 8-hydroxyguanosine (8-OHdG, marker of oxidative stress to DNA) [9], protein carbonyls (markers of protein oxidation) [10], lipid-peroxidation products [11,12], and advanced lipoxidation end-products [13]. The cerebrospinal fluid, a fluid in close contact with neural tissue and good witness of neurodegeneration processes, also shows signs of peroxidation in PD. The enzyme DJ-1 (PARK7), reported to act as an antioxidant peroxidase, is reduced in CSF in the initial phases of PD [14]. Buhmann et al. [15] detected enhanced lipid peroxidation in the CSF, and high levels of malondialdehyde (MDA), a compound that causes toxic stress in cells, have been reported too [16,17]. Other markers for oxidative stress such as the oxidized forms of coenzyme-Q and 8-OHdG are augmented in CSF of PD patients [18]. The anti-oxidant ferroxidase activity of ceruloplasmin is also reduced in the CSF of PD [19-21]. Regarding anti-oxidant molecules encompassing the glutathione (GSH) system, several studies have indicated that levels of total glutathione protein are diminished in CSF and brain tissue of PD [22-24], although data on glutathionerelated enzymes are contradictory [25-27]. The blood of patients with PD also show signs of peroxidation stress, because it has been detected increase of 8-OHdG, MDA, and vitamin E, and reduction of the antioxidant enzyme GPx [16,28].
My research group has detected a strong peroxidation stress in CSF of patients with PD because the enzymatic activity of the three principal hydrogen peroxide scavengers is reliably reduced [29]. Thus after analyzing the activity of superoxide anion and hydrogen peroxide (H2O2) scavengers, the data revealed a disturbance of H2O2 scavenging, but not superoxide anion scavenging, as manifested through a significantly reduced activity of GPx, catalase, and PRDxs, without changes in the activity of SODs [29]. GPx are selenoproteins that reduce peroxides in water. They also reduce lipid hydroperoxides to their corresponding alcohols. Catalases are ubiquitous enzymes that mediate the decomposition of H2O2 in water, as explained. PRDxs are cysteine-dependent peroxidases that react with hydrogen peroxide and hydroperoxide substrates. The reliably reduction of the activity of hydrogen peroxide scavengers is expected to activate alternative pathways in order to scavenge H2O2. These pathways include the Fenton reaction, the Haber-Weiss reaction, and the formation of hypohalous acids after activation of haloperoxidases. In addition, dopamine catabolism of dopaminergic neurons, cells that die in the course of PD, leads to either the formation of H2O2 under the action of monoamino-oxidases (MAO), or the generation of dopamine-quinones [30]. All of these pathways, even though they facilitate the elimination of H2O2, give to the formation of radical species and quinones, highly oxidant molecules, which would worsen oxidative stress [30]. Figure 2 shows these alternative pathways that are activated after failure of normal H2O2 scavenging, and that are linked to peroxidation stress in PD. Our observation of reduced H2O2 scavenging in the CSF of patients leads to the hypothesis that the activation of these alternative pathways is a critical event in the parkinsonian central nervous system. In this context, ferritin levels and iron storage have also been observed to be altered in CSF and central nervous system of patients, and it is well known that the enhancement of transition metals such as iron and cupper facilitates the Fenton reaction to occur [30].
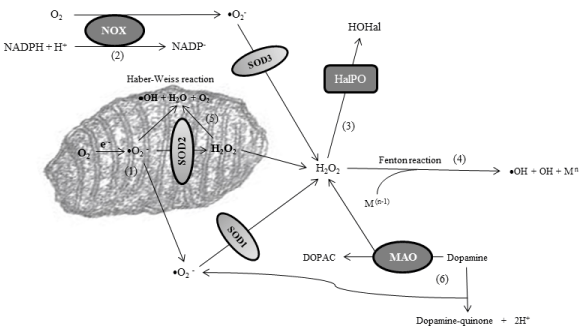
Figure 2: Alternative pathways that are activated after failure of normal H2O2 scavenging. As explained before, the main production of superoxide anion (•O2
−) is
in the mitochondrial matrix [1] or through the NADPH-oxidase (NOX) enzymes [2]. Superoxide anions are catalyzed into hydrogen peroxide through superoxide
dismutases (SODs). The excess of hydrogen peroxide o its inadequate scavenging leads to the action of haloperoxidases (HalPO), enzymes which catalyze
the formation of hypohalous acids (HOHal) [3], as well as to the activation of the Fenton reaction if free transition metals (with [n-1] electronic structure) are also
enhanced [4]. The Fenton reaction leads to the formation of reactive species such as hydroxyl ion (•OH). In addition, excess of superoxide anion and H2O2 inside
the mitochondria activates the Haber-Weiss reaction, another source of reactive species [5]. Finally, in dopaminergic neurons, cells that die in the course of PD,
there is overproduction of H2O2 (under the action of monoamino-oxidases or MAO), dopamine-quinones and •O2
−, all worsening oxidative stress [6].
Misfolding of proteins is another deleterious effect of peroxidation stress, which alters normal protein function and/or facilitates the formation of proteinaceous aggregates inside neurons. Quinones, which are also formed during peroxidation stress in dopamine neurons in PD, also contribute to misfolding of proteins due to the formation of protein cross-linking [30]. Several of these misfolded proteins such as α-synuclein (α-SYN) and parkin (PARK2) are classically associated to the pathogenesis of PD. Misfolded α-SYN tends to form protofibrils which would precipitate forming fibrils which in turn constitute the core of the Lewy bodies (LBs), anatomo-pathological hallmarks of PD. Protofibrils have been shown to be the neurotoxic species of α-SYN [31]. Association of misfolded α-SYN with peroxidized lipid metabolites can lead to mitochondrial dysfunction that in turn leads to dopaminergic neuron death [31]. Peroxidation stress is also known to induce structural changes in α-SYN leading to covalent aggregation [32]. Regarding Parkin (PARK2), this protein is an E3 ubiquitin lygase involved in the ubiquitination of proteins, and this activity is inhibited by peroxidation. The inhibition of parkin function could contribute to the degenerative process by impairing the ubiquitination of parkin substrates [33]. Parkin is also a transcriptional repressor of p53 [34], and peroxidation alters this function that is also associated with PD [35]. It is worth recalling that recessive mutations in the PARK2 gene encoding Parkin are responsible of many juvenile and early-onset cases of parkinsonism [36].
Halogenation and Parkinson’s disease
Halogenation stress is directly associated to peroxidation stress, because overproduction of hydrogen peroxide or its reduced scavenging leads to the activation of halogenation pathways (Figure 2). The role of halogenation stress in PD has not been discerned, but likely it could play a significant role in the pathogenesis of PD [37,38]. Halogenation reactions are mediated by haloperoxidases, which encompass several enzymes such as white blood-cells peroxidases (myeloperoxidase or MPO, eosinophil-peroxidase or EPO), and glandular haloperoxidases (thyroperoxidase or TPO; lactoperoxidase or LPO) [5,6]. Haloperoxidases mediate the conversion of hydrogen peroxide and halogens into hypohalous acids, such as hypochlorous or hypoiodous acids. As explained, the physiological formation of hypohalous acids and haloamines is mostly scavenged by glutathione- S-transferase and sulfotransferases [7]. However if there is excess of H2O2, as it seems to be the case in PD [1-4], hypohalous acids can lead to the overproduction of haloamines and halogenated proteins. Among haloamines, those derived from tyrosine are of high interest for understanding PD, because halotyrosines include molecules such as chlorotyrosines and iodotyrosines that are considered as neurotoxic for dopamine neurons [37,39]. Halogens can be incorporated at position 3 and 5 of the 6-carbon aromatic ring of tyrosine, leading to the formation of 3-monohalotyrosines or 3,5-dihalotyrosines. As for halogenated proteins, they are gaining increased importance in PD, because they can act as proinflammatory mediators. For instance, chlorinated proteins originate as a result of the action of free radicals such as chloramines and hypochlorous acid on proteins, and they act as inflammatory mediators after binding albumin [40-42]. Of note is that characterizing protein markers associated with halogenation stress is of great interest for biomedicine, because proteins or adducted protein products are good candidates for biomarkers since they show great stability, early formation, and longer lifespan [43]. The molecular pathways which are involved in the formation and degradation of haloamines and halogenated proteins are illustrated in Figure 3.
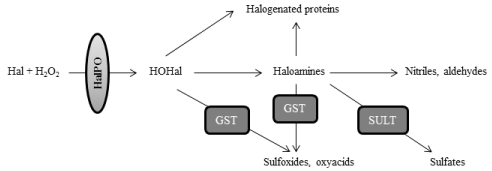
Figure 3: Molecular pathways which are involved in the formation and degradation of haloamines and halogenated proteins. Haloperoxidases (HalPO) mediate the
conversion of hydrogen peroxide into hypohalous acids (HOHal), after the incorporation of halogens (Hal). Hypohalous acids can react with primary amines to form
haloamines. Hypohalous acids and haloamines are scavenged by glutathione-S-transferase, leading to the formation of oxyacids and sulfoxides. Haloamines can
also be eliminated by sulfation through the action of sulfotransferases (SULT), or after the formation of nitriles and aldehydes, although these latter reactions are
slow in normal physiology. If there is peroxidation stress and excess of H2O2, the enhanced formation of hypohalous acids and halogenated amines would lead to
the formation of halogenated proteins, due to the incorporation of halogens on proteins.
Among halogenated proteins, advanced oxidation protein products (AOPP) are considered as promising markers for halogenation stress [37,40]. These protein products originate as a result of the action of halogenation radicals on proteins, and they are known to be augmented in serum and cerebrospinal fluid of PD patients [37]. Hypohalous acids include hypochlorous (HOCl), hypobromous (HOBr) and hypoiodous acids (HOI), leading to chlorinated, brominated and iodinated AOPP, respectively [40,41,42,44,45]. As shown in Figure 3, haloperoxidases catalyze these reactions leading to hypohalous acids from halogens and hydrogen peroxide. AOPP can conjugate with human serum albumin (HSA) giving AOPP-HSA conjugates. Of note is that AOPP-HSA conjugates can act as proinflammatory factors, because chlorinated AOPP-HSA are known to be inflammatory mediators triggering the oxidative “ignition” of neutrophils, monocytes and T-lymphocytes [46,47]. Chlorinated AOPP are enhanced in many inflammatory diseases, and they correlate with several measures of these diseases [44,46-48]. Hence halogenated protein products could be linked to initial inflammatory processes in PD, where immunological and inflammatory responses in the vicinity of dopaminergic neurons have been detected [49,52].
Our research group has demonstrated that AOPP are enhanced in serum and CSF of parkinsonian patients [37]. Serum concentration of these protein products is progressively reduced over time, and levodopa treatment contributes to this reduction [37], although there is no a statistical relationship because only 47% of our patients received levodopa. In addition, after analyzing the correlation between duration of the disease and serum AOPP levels in patients with moderate PD, a significant correlation was found (p <0.003). In other words, low serum AOPP levels can predict a longer duration of the disease and a longer duration in reaching advanced stages of the disease, and more than 80% of studied patients with a PD longer than 10 years had serum AOPP levels lower than 350 μM (normal value in control subjects is 148.4±37 μM) [37].
Serum level of advanced oxidation protein products could hence be a prognostic marker of duration of PD. However, specificity of AOPP as a PD biomarker is low because these products also accumulate in other diseases such as diabetes mellitus, uremic syndrome, atherosclerosis, systemic sclerosis and acquired immune deficiency syndrome (AIDS) [53-58]. Then it is urgently needed to identify the specific molecular pathways which are involved in AOPP synthesis in PD. As explained, halogenation stress and AOPP formation are associated to the action of haloperoxidases, and different haloperoxidases mediate different halogenation processes. MPO (haloperoxidase of white blood cells) mediates the formation of hypochlorous and hypobromide acids, with leads to the formation of AOPP with chloride and bromide radicals. The MPO-related pathway is the most activated in above cited diseases, but MPO seems not to be involved in the formation of AOPP in PD, because this enzyme is found not to be enhanced in serum or CSF of PD patients [59]. TPO, haloperoxidase of thyroid origin involved in iodide management, is found to be elevated in more than 25% of patients with PD [59]. TPO mediates the formation of hypoiodous acid, which leads to the production of iodinated AOPP. It seems plausible that serums AOPP in PD are iodinated protein products, and they could exert proinflammatory effects. In this context, the thyroid gland has been linked to PD, because some researchers have found more prevalence of some thyroid dysfunctions among PD patients or they have detected sympathetic denervation of the thyroid gland in PD patients, a hallmark of the disease because it also occurs in other organs such as heart, stomach and rectum [60-63]. Finally, the role of other human haloperoxidases such as EPO and LPO has not been studied. EPO is detected in serum and CSF like MPO, and it is involved in the formation of hypochlorous and hypobromide acids. LPO is detected in saliva and milk, and it is mostly involved in the formation of hypoiodous and hypobromide acids.
Conclusion
Oxidative stress, which is defined as an imbalance between the production of reactive oxidative species and anti-oxidant mechanisms, is considered as an important pathogenic mechanism in PD. Two types of oxidative stress, peroxidation and halogenation stress, are gaining increasing importance as biochemical windows to a better understanding of the pathogenesis of this disease. Peroxidation stress is associated with excess of hydrogen peroxide, and it induces the presence of many peroxidation-related molecular markers in Parkinsonian patients, and to misfolding of proteins. Many of these proteins, such as α-synuclein and parkin, are traditionally linked to PD. Peroxidation-induced misfolded proteins show altered functionality, such as loss of neuroprotective activity or the tendency to form proteinaceous aggregates in neurons. This latter effect could account for the formation of Lewy bodies, anatomo-pathological hallmark of PD. Peroxidation stress is also detected by a loss of the activity of the main hydrogen peroxide scavengers, glutathioneperoxidase, catalase, and peroxiredoxins, in the cerebrospinal fluid of patients. Altered hydrogen peroxide scavenging leads to formation of oxidative radicals, alteration in iron storage, and halogenation stress. Halogenation stress is characterized by the excess of halogenated molecules such as hypohalous acids, haloamines and halogenated proteins. Among halogenated proteins, advanced oxidation protein products or AOPP are augmented in Parkinson’s disease. Halogenated amines and proteins are thought to be deleterious for neurons, and the author hypothesizes that they could play a critical role in the ethiology and development of Parkinson’s disease.
Acknowledgement
The author thanks Mara Guerra and Silvia Castellano (BIO127 lab, University of Seville) for their excellent technical assistance; Drs. Angel Martín de Pablos (Surgery, University of Seville), José-Manuel García-Moreno, Fátima Damas-Hermoso (Neurology, Hospital Macarena Seville), and José Chacón (Neurology, Hospital Infanta Luisa, Seville) for their excellent collaboration; and Dr. Fernando Rodriguez de Fonseca (Instituto de Investigación Biomédica de Málaga) for his friendship and help. Supported by grants to EF from Junta de Andalucia (BIO127).
References
- Fahn S, Cohen G. The oxidant stress hypothesis in Parkinson’s s disease: evidence supporting it. Ann Neurol. 1992; 32: 804-812.
- Jenner P. Oxidative stress in Parkinson's disease. Ann Neurol. 2003; 53: S26-36.
- Schapira AH, Olanow CW, Greenamyre JT, Bezard E. Slowing of neurodegeneration in Parkinson's disease and Huntington's disease: future therapeutic perspectives. Lancet. 2014; 384: 545-555.
- Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinson Dis. 2013; 3: 461-491.
- Dunforb HB. Myeloperoxidase and eosinophil peroxidase: phagocytosis and microbial killing. In: Dunforb HB, editor. Heme Peroxidases, Wiley. 1999; 349-385.
- Harrison JE, Schultz J. Studies on the chlorinating activity of myeloperoxidase. J Biol Chem. 1976; 251: 1371-1374.
- Yasuda S, Yasuda T, Liu MY, Shetty S, Idell S, Boggaram V, et al. Sulfation of chlorotyrosine and nitrotyrosine by human lung endothelial and epithelial cells: role of the human SULT1A3. Toxicol Appl Pharmacol. 2011; 251: 104-109.
- Mani AR, Ippolito S, Moreno JC, Visser TJ, Moore KP. The metabolism and dechlorination of chlorotyrosine in vivo. J Biol Chem. 2007; 282: 29114-29121.
- Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, et al. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997; 69: 1196-1203.
- Floor E, Wetzel MG. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J Neurochem. 1998; 70: 2682-2675.
- Dexter D, Carter C, Agid F, Agid Y, Lees AJ, Jenner P, et al. Lipid peroxidation as cause of nigral cell death in Parkinson's disease. Lancet. 1986; 2: 639-640.
- Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, et al. Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease. J Neurochem. 1989; 52: 381-389.
- Castellani R, Smith MA, Richey PL, Perry G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 1996; 737: 195-200.
- Hong Z, Shi M, Chung KA, Quinn JF, Peskind ER, Galasko D, et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson's disease. Brain. 2010; 133: 713-726.
- Buhmann C, Arlt S, Kontush A, Möller-Bertram T, Sperber S, Oechsner M, et al. Plasma and CSF markers of oxidative stress are increased in Parkinson's disease and influenced by antiparkinsonian medication. Neurobiol Dis. 2004; 15: 160-170.
- Ilic TV, Jovanovic M, Jovicic A, Tomovic M. Oxidative stress indicators are elevated in de novo Parkinson's disease patients. Funct Neurol. 1999; 14: 141-147.
- Pryor WA, Stanley JP. Letter: A suggested mechanism for the production of malonaldehyde during the autoxidation of polyunsaturated fatty acids. Nonenzymatic production of prostaglandin endoperoxides during autoxidation. J Org Chem. 1975; 40: 3615-3617.
- Isobe C, Abe T, Terayama Y. L-Dopa therapy increases homocysteine concentration in cerebrospinal fluid from patients with Parkinson's disease. J Clin Neurosci. 2010; 17: 717-721.
- Boll MC, Sotelo J, Otero E, Alcaraz-Zubeldia M, Rios C. Reduced ferroxidase activity in the cerebrospinal fluid from patients with Parkinson's disease. Neurosci Lett. 1999; 265: 155-158.
- Boll MC, Alcaraz-Zubeldia M, Montes S, Rios C. Free copper, ferroxidase and SOD1 activities, lipid peroxidation and NO(x) content in the CSF. A different marker profile in four neurodegenerative diseases, Neurochem Res. 2008; 33: 1717-1723.
- Abdi F, Quinn JF, Jankovic J, McIntosh M, Leverenz JB, Peskind E, et al. Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neurodegenerative disorders. J Alzheimers Dis. 2006; 9: 293-348.
- Perry TL, Godin DV, Hansen S. Parkinson's disease: a disorder due to nigral glutathione deficiency? Neurosci Lett. 1982; 33: 305-310.
- Fitzmaurice PS, Ang L, Guttman M, Rajput AH, Furukawa Y, Kish SJ. Nigral glutathione deficiency is not specific for idiopathic Parkinson's disease. Mov Disord. 2003; 18: 969-976.
- Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, et al. Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994; 36: 348-355.
- Kish SJ, Morito C, Hornykiewicz O. Glutathione peroxidase activity in Parkinson's disease brain. Neurosci Lett. 1985; 58: 343-346.
- Damier P, Hirsch E, Javoy-Agid F. Glutathione peroxidase, glial cells and Parkinson´s disease. Neuroscience. 1993; 86: 321-331.
- Guo J, Sun Z, Xiao S, Liu D, Jin G, Wang E. Proteomic analysis of the cerebrospinal fluid of Parkinson's disease patients. Cell Res. 2009; 19: 1401-1403.
- Chen CM, Liu JL, Wu YR, Chen YC, Cheng HS, Cheng ML, et al. Increased oxidative damage in peripheral blood correlates with severity of Parkinson's disease. Neurobiol Dis. 2009; 33: 429-435.
- Martín de Pablos A, García-Moreno JM, Fernández E. Does the Cerebrospinal Fluid Reflect Altered Redox State But Not Neurotrophic Support Loss in Parkinson's Disease? Antioxid Redox Signal. 2015; 23: 893-898.
- Navarro-Yepes J, Burns M, Anandhan A, Khalimonchuk O, del Razo LM, Quintanilla-Vega B, et al. Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid Redox Signal. 2014; 21: 66-85.
- Ruipérez V, Darios F, Davletov B. Alpha-synuclein, lipids and Parkinson's disease. Prog Lipid Res. 2010; 49: 420-428.
- Olteanu A, Pielak GJ. Peroxidative aggregation of alpha-synuclein requires tyrosines. Protein Sci. 2004; 13: 2852-2856.
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004; 304: 1328-1331.
- Alves da Costa C, Checler F. Apoptosis in Parkinson's disease: is p53 the missing link between genetic and sporadic Parkinsonism? Cell Signal. 2011; 23: 963-968.
- Meng F, Yao D, Shi Y, Kabakoff J, Wu W, Reicher J, et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol Neurodegener. 2011; 6: 34.
- Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006; 7: 306-318.
- García-Moreno JM, Martín de Pablos A, García-Sánchez MI, Méndez-Lucena C, Damas-Hermoso F, Rus M, et al. May serum levels of advanced oxidation protein products serve as a prognostic marker of disease duration in patients with idiopathic Parkinson's disease? Antioxid Redox Signal. 2013; 18: 1296-1302.
- Yap YW, Whiteman M, Cheung NS. Chlorinative stress: an under appreciated mediator of neurodegeneration? Cell Signal. 2007; 19: 219-228.
- Scholz J, Toska K, Luborzewski A, Maass A, Schünemann V, Haavik J, et al. Endogenous tetrahydroisoquinolines associated with Parkinson's disease mimic the feedback inhibition of tyrosine hydroxylase by catecholamines. FEBS J. 2008; 275: 2109-2121.
- Witko-Sarsat V, Gausson V, Nguyen AT, Touam M, Drüeke T, Santangelo F, et al. AOPP-induced activation of human neutrophil and monocyte oxidative metabolism: a potential target for N-acetylcysteine treatment in dialysis patients. Kidney Int. 2003; 64: 82-91.
- Dunforb HB. Myeloperoxidase and eosinophil peroxidase: phagocytosis and microbial killing. In: Dunforb HN, Editor. Heme Peroxidases. Wiley. 1999; 349-385.
- Yap YW, Whiteman M, Cheung NS. Chlorinative stress: an under appreciated mediator of neurodegeneration? Cell Signal. 2007; 19: 219-228.
- Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta. 2003; 329: 23-38.
- Curtis MP, Hicks AJ, Neidigh JW. Kinetics of 3-chlorotyrosine formation and loss due to hypochlorous acid and chloramines. Chem Res Toxicol. 2011; 24: 418-428.
- Harrison JE, Schultz J. Studies on the chlorinating activity of myeloperoxidase. J Biol Chem. 1976; 251: 1371-1374.
- Descamps-Latscha B, Witko-Sarsat V. Importance of oxidatively modified proteins in chronic renal failure. Kidney Int Suppl. 2001; 78: S108-113.
- Robaszkiewicz A, Bartosz G, Soszyński M. N-chloroamino acids cause oxidative protein modifications in the erythrocyte membrane. Mech Ageing Dev. 2008; 129: 572-579.
- Buss IH, Senthilmohan R, Darlow BA, Mogridge N, Kettle AJ, Winterbourn CC. 3-Chlorotyrosine as a marker of protein damage by myeloperoxidase in tracheal aspirates from preterm infants: association with adverse respiratory outcome. Pediatr Res. 2003; 53: 455-462.
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988; 38: 1285-1291.
- Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debré P, et al. FcepsilonRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci. 1999; 19: 3440-3447.
- Hunot S, Hirsch EC. Neuroinflammatory processes in Parkinson's disease. Ann Neurol. 2003; 53: S49-58.
- Nagatsu T, Sawada M. Cellular and molecular mechanisms of Parkinson's disease: neurotoxins, causative genes, and inflammatory cytokines. Cell Mol Neurobiol. 2006; 26: 781-802.
- Witko-Sarsat V, Friedlander M, Capeillère-Blandin C, Nguyen-Khoa T, Nguyen AT, Zingraff J, et al. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996; 49: 1304-1313.
- Kalousová M, Skrha J, Zima T. Advanced glycation end-products and advanced oxidation protein products in patients with diabetes mellitus. Physiol Res. 2002; 51: 597-604.
- Witko-Sarsat V, Nguyen Khoa T, Jungers P, Drüeke T, Descamps-Latscha B. Advanced oxidation protein products: oxidative stress markers and mediators of inflammation in uremia. Adv Nephrol Necker Hosp. 1998; 28: 321-341.
- Malle E, Waeg G, Schreiber R, Gröne EF, Sattler W, Gröne HJ. Immunohistochemical evidence for the myeloperoxidase/H2O2/halide system in human atherosclerotic lesions: colocalization of myeloperoxidase and hypochlorite-modified proteins. Eur J Biochem. 2000; 267: 4495-4503.
- Gröne HJ, Gröne EF, Malle E. Immunohistochemical detection of hypochlorite-modified proteins in glomeruli of human membranous glomerulonephritis. Lab Invest. 2002; 82: 5-14.
- Bräsen JH, Häkkinen T, Malle E, Beisiegel U, Ylä-Herttuala S. Patterns of oxidized epitopes, but not NF-kappa B expression, change during atherogenesis in WHHL rabbits. Atherosclerosis. 2003; 166: 13-21.
- Fernández E, García-Moreno JM, Martín de Pablos A, Chacón J. May the thyroid gland and thyroperoxidase participate in nitrosylation of serum proteins and sporadic Parkinson's disease? Antioxid Redox Signal. 2014; 21: 2143-2148.
- García-Moreno JM, Chacón J. [Hypothyroidism concealed by Parkinson's disease]. Rev Neurol. 2002; 35: 741-742.
- Matsui H, Udaka F, Oda M, Tamura A, Kubori T, Nishinaka K, et al. Metaiodobenzylguanidine (MIBG) uptake in Parkinson's disease also decreases at thyroid. Ann Nucl Med. 2005; 19: 225-229.
- Tandeter H, Levy A, Gutman G, Shvartzman P. Subclinical thyroid disease in patients with Parkinson's disease. Arch Gerontol Geriatr. 2001; 33: 295-300.
- Tipre DN, Goldstein DS. Cardiac and extracardiac sympathetic denervation in Parkinson's disease with orthostatic hypotension and in pure autonomic failure. J Nucl Med. 2005; 46: 1775-1781.