
Review Article
Austin J Clin Neurol 2016; 3(1): 1086.
Facilitation of the Brain Hepatocyte Growth Factor/ C-Met Receptor System: A New Approach to Treat Alzheimer’s Disease?
Wright JW*, Kawas LH and Harding JW
Departments of Psychology, Integrative Physiology and Neuroscience, Washington State University, USA
*Corresponding author: John W. Wright, Department of Psychology, Integrative Physiology and Neuroscience, Washington State University, P.O. Box 644820, Pullman, WA 99164-4820, USA
Received: March 30, 2016; Accepted: May 13, 2016; Published: May 15, 2016
Abstract
Alzheimer’s disease (AD) is a major neurodegenerative disorder presently without adequate treatment that is increasing in frequency as life expectancy increases. New therapeutic approaches are needed to slow and hopefully reverse disease progression. Neurotrophic agents such as nerve growth factor and brain-derived neurotrophic factor have received research attention concerning their potential to treat AD but have not progressed to clinical trials due to their reasonably large size, inability to penetrate the blood-brain barrier (BBB), and the high cost of synthesis. This review focuses on one over looked neuro trophin, hepatocyte growth factor (HGF) that acts via the Type 1 tyrosine kinase receptor c-Met to mediate stem cell differentiation, synaptogenesis, neurogenesis, and protect against tissue insults in a wide range of cell types including neurons. We have determined that the brain angiotensin and HGF/c- Met systems interact in such a way that angiotensin IV (Ang IV)-based analogs including Nle¹-AngIV, Dihexa and others stimulate HGF dimerization which is a prerequisite to binding at the c-Met receptor. These analogs have shown the ability to facilitate the formation of new functional synaptic connections in hippocampal slices, promote neurogenesis, and augment memory consolidation and retrieval in animal models of AD. This family of compounds represents a new class of drugs with lead candidates that are orally active, penetrate the BBB sufficiently to reach therapeutic concentrations, and reverse memory deficits seen in animal models of dementia.
Keywords: Alzheimer’s disease, Angiotensin IV, Nle¹-Angiotensin IV, Dihexa, AT4 receptor subtype, Hepatocyte growth factor, c-Met receptor
Abbreviations
Aβ: Amyloid Beta Protein; ACE: Angiotensin Converting Enzyme; Ach: Acetylcholine; ACSF: Artificial Cerebrospinal Fluid; AD: Alzheimer’s Disease; Ang: Angiotensin; Ang(3-7): Angiotensin II(3-7); AngI: Angiotensin I; AngII: Angiotensin II; AngIII: Angiotensin III; AngIV: Angiotensin IV; AP-A: Aminopeptidase A; AP-N: Aminopeptidase N; ARBs: Angiotensin Receptor Blockers; AT1: Angiotensin Receptor Subtype 1; AT2: Angiotensin Receptor Subtype 2; AT4: Angiotensin Receptor Subtype 4; BBB: Blood- Brain Barrier; BDNF: Brain-Derived Neurotrophic Factor; Carb-P: Carboxypeptidase P; CBF: Cerebral Blood Flow; CIP: Chromatin Immunoprecipitation; c-Met: Met Type 1 Receptor Tyrosine Kinase; D: Aspartate; ERK: Extracellular Signaling-Regulated Kinase; F: Phenylalanine; FDA: Federal Drug Administration; H: Histidine; HGF: Hepatocyte Growth Factor; HIV: Human Immunodeficiency Virus; I: Isoleucine; Ile: Isoleucine; K: Lysine; L: Leucine; LTP: Long-Term Potentiation; LVV-H7: Leucine-Valine-Valine- Hemorphin-7; MAPK: Mitogen Activated Protein Kinase; MCI: Mild Cognitive Impairment; N: Asparagine; NGF: Nerve Growth Factor; Nle: Norleucine; NMDA: N-Methyl-D-Aspartate; NO: Nitric Oxide; NSAIDs: Non-Steroidal Anti-Inflammatory Drugs; NT3: Neurotrophin-3; NT4: Neurotrophin-4; P: Proline; Phe: Phenylalanine; ψ: CH2-NH2; PO: Propyl Oligopeptidase; Pro: Proline; P13K: Phosphatidylinositol 3-Kinase; R: Arginine; RAS: Renin- Angiotensin System; SK: Scatter Factor; SPH: Serine Proteinase Homology; Tyr: Tyrosine; V: Valine; Y: Tyrosine
Introduction
Alzheimer’s disease (AD) is characterized by elevated levels of amyloid plaques and neurofibrillary tangles that predispose progressive neuron losses in memory related structures including neocortex, piriform cortex, hippocampus and the nucleus basalis of Meynert [1,2]. AD currently afflicts approximately 5.3-6 million Americans with annual treatment and care costs estimated at $70-100 billion [3,4]. These patients respond only marginally to presently available FDA approved drugs [5,6]. In the absence of a breakthrough in treatment the number of AD patients is predicted to reach 16 million in the U.S. by mid-century with associated health care costs in excess of $500 billion [4,7]. Such costs will cripple our health care system. The goal of providing an effective treatment for AD has been elusive due to the complexity of the disease process and resulting inability to identify reliable biomarkers. In addition, AD diagnostic indicators are present in other clinical conditions including vascular disease, frontotemporal dementia, Parkinson’s disease and HIV infection induced dementia, as well as normal aging [8-11]. These considerations make drug development to treat AD a very challenging task. A treatment designed to delay the onset of symptoms would prolong and maintain the patient’s quality of life and significantly reduce health care costs. De la Torre [12] has calculated that postponing the onset of AD by 5 years could reduce patient numbers by upwards of 50%. Recently it has been reported that the presence of two positive biomarkers for AD, β-amyloid and neuro degeneration, and the use of in vivo amyloid imaging agents, offer pre-diagnostic predictive value regarding the trajectory of cognitive change [13,14]. These findings promise to be of major importance regarding diagnosis but not prevention of AD.
Current drugs to treat Alzheimer’s disease
Available FDA approved drugs to treat AD fall into two major classes (Table 1): 1) Namenda (memantine HCl) acts as an N-methyl-D- aspartate (NMDA) receptor antagonist designed to limit glutamate excitotoxicity and resulting neuronal damage [15-17]. Namenda has shown positive results in some patients particularly if given in combination with acetylcholinesterase inhibitors [5,6]. 2) Cholinesterase inhibitors such as Razadyne, Exelon, Cognex and Aricept disrupt the degradation of acetylcholine (Ach) thus extending the half-life and availability of this neurotransmitter acting at central cholinergic muscarinic and nicotinic receptors. Additional treatment approaches being vigorously pursued include monoclonal antibodies designed to attenuate and block the production and deposition of insoluble amyloid β (Aβ) protein fragments resulting from amyloid precursor protein proteolysis. It is suggested that dysfunction between Aβ production and clearance causes damaging accumulations of cellular Aβ, coupled with hyper phosphorylation of neuronal tau protein resulting in neurofibrillary tangle formation [18-20]. Antiinflammatories and anti-oxidants are also being tested including nonsteroidal anti-inflammatory drugs (NSAIDs, eg. naproxen, rofecoxib, ibuprofen, indomethacin, tarenflurbil, diclofenac/misoprostol), luteolin, ferulic acid to protect against neurotoxicity [21-24].
![]()
Drug/Compound
Mechanism of Action
Clinical Target
Reference
Namenda
NMDA receptor antagonist
Mid-stage AD
[15-17]
Cholinesterase inhibitors
Interfere with degradation of Ach
Early- mid-stage AD
[5,6]
Monoclonal antibodies against Aβ
Block intracellular accumulation of Aβ1-42
Early-stage AD
[18-20]
Anti-inflammatories
Reduce inflammation, promote neuro protection
Early- mid-stage AD
[21-24]
Nle1-AngIV, Dihexa
Increase HGF dimerization and facilitate binding to c-Met
Proposed early-stage AD
[45,81,93]
Norleual-AngIV
Reduce HGF dimerization and inhibit binding to c-Met
Proposed treatment against carcinomas
[29,30,78]
Table 1: FDA approved drugs to treat Alzheimer’s disease and AngIV-based analogs that interact with the HGF/c-Met receptor system.
An interim treatment strategy designed to offset neuron losses by stimulating synaptogenesis in existing neurons and the formation of new functional neurons would be advantageous in slowing disease progression. The neurotrophic agents capable of facilitating synaptogenesis and neurogenesis include nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3, and neurotrophin-4 [25,26]. To date BDNF has received the most attention [27]. Our laboratory has focused on an overlooked neurotrophic factor, hepatocyte growth factor (HGF), and found it to be more potent than BDNF when activated by angiotensin IV (AngIV)-based analogs [28]. These analogs allosterically mimic dimerization/activation, a prerequisite to binding to the Type 1 tyrosine kinase receptor c-Met [29,30]. This review initially describes the renin-angiotensin system’s (RAS) role in memory formation followed by descriptions of neurotrophic agents and the HGF/c-Met system. We conclude with details concerning the development and testing of AngIV-based analogs that activate the brain HGF/c-Met receptor system and show promise as anti-dementia agents in animal models of AD. We also detail the limitations of these molecules.
The renin-angiotensin system and memory
The classic RAS is recognized for its role in regulating blood pressure and body water balance as mediated by the octapeptide angiotensin II (AngII) acting at the G-protein coupled AT1 receptor subtype(40-42 kDa; [31]). This AngII/AT1 receptor system has been a major focus regarding the development of antihypertensive drugs and its role in inflammation, oxidative stress and tissue remodeling [32,33]. These latter processes contribute to the “neuronal inflammation response” a key factor in neurodegenerative diseases including AD [34-36]. A role for AngII in memory was suggested some time ago focused on AngII interacting with the AT1 receptor subtype (reviewed in [2,37-39]). More recently members of our laboratory discovered the AT4 receptor protein (160-190 kDa) and the importance of the hexapeptide AngIV acting at this receptor subtype in the facilitation of memory acquisition and retrieval [28,40,41]. Subsequent findings indicated that the learning and memory enhancing effects originally attributed to AngII acting at the AT1 receptor were due to the enzymatic conversion of AngII to AngIII and then to AngIV acting at the AT4 receptor (Figure 1) [42-44]. It is now clear that AngII interferes with performance by animal models of AD on most memory tasks while AngIV facilitates performance.
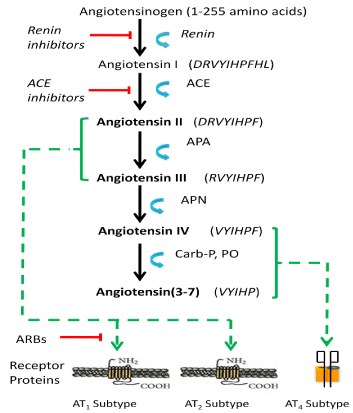
Figure 1: Angiotensin structures and synthesis pathway to produce
biologically active ligands. The angiotensin synthesis pathway is presented
indicating biologically active ligands (bold font), enzymes, and receptors
involved in mediating related physiologies and behaviors. Angiotensinogen
is converted to the decapeptide angiotensin I by renin. Angiotensin I is
converted to the octapeptide angiotensin II by angiotensin converting
enzyme (ACE). Aminopeptidase A (APA) converts angiotensin II to the
heptapeptide angiotensin III that is converted to the hexapeptide angiotensin
IV by aminopeptidase N (APN). Angiotensins II and III bind at the AT1and
AT2receptor subtypes; while angiotensin IV binds at the AT4 receptor
subtype. Antihypertensive drugs such as ACE inhibitors act to interfere with
conversion of angiotensin I to the biologically active form angiotensin II, and
angiotensin receptor blockers (ARBs) act as antagonists at the AT1 receptor
thus preventing angiotensin II binding.
The analogNle¹-AngIV (Norleucine-YIHPF) overcomes the memory impairments evidenced by animal models of AD. Specifically, intracerebroventricular treatment with Nle¹-AngIV reverses memory deficits due to: 1) application of the cholinergic muscarinic receptor antagonist scopolamine; 2) kainic acid-induced lesions of the hippocampus; 3) perforant path knife-cuts; 4) embolic stroke due to carotid artery injection of microspheres; 5) treatment with the angiotensin receptor blocker (ARB) losartan; and 6)ischemia resulting from transient four-vessel occlusion (reviewed in [45]). This latter finding is particularly important given the possibility that cerebral hypoperfusion may act as a precursor to the development of mild cognitive impairment (MCI), a condition that often precedes the onset of AD [46]. To date we have not tested an AngIV-based analog for efficacy in a transgenic mouse model of AD. Consistent with the above behavioral results [125I] AngIV has been auto radiographically localized within structures known to mediate cognitive processing including human neocortex, hippocampus, and the basal nucleus of Meynert [2,47].
Neurotrophic agents
Over the past 60 years several neurotrophic agents have been identified in the mammalian brain and their roles in neurogenesis, neurite outgrowth and neural protection have been studied (reviewed in [48]). Nerve growth factor (NGF) was the first to be discovered [49] followed by the purification of BDNF [50]. Most recently neurotrophin-3 (NT3) and neurotrophic-4 (NT4) have been isolated in the mammalian brain; while HGF was first isolated from the liver [51] and has now been identified in the brain [52-54].
Neurotrophic agents promote brain neuronal survival while decreases in their levels have been measured in several neurodegenerative diseases [55,56]. Thus, neurotrophins have been suggested as potential treatments against AD, Parkinson’s disease and other neurodegenerative diseases [57]. BDNF has received the most attention due to its importance in the development and ongoing maintenance of normal brain functioning. Significant declines in BDNF expression have been seen in several neurodegenerative diseases [58-60] and BDNF has been implicated as important in Alzheimer’s [61] and Huntington’s diseases [62]. BDNF has been shown to overcome learning deficits in animal models of AD [63], and reveals increased expression with treadmill exercise [64]. NGF is synthesized in the hippocampus and neocortex and is transported to cholinergic neurons located in the forebrain. Since AD patients suffer from deficits in axonal transport NGF has been suggested as a treatment strategy [65,66]. Somewhat less attention has been given to NT3 and NT4, although their roles as brain neurotrophic agents are important (reviewed in [67-69].
The hepatocyte growth factor/c-Met receptor system
The plasminogen family member HGF, also known as “scatter factor”, has been shown to promote liver regeneration [52-54]. HGF acts at the c-Met receptor to stimulate mitogenesis, motogenesis and morphogenesis in a number of cellular targets including epithelial, endothelial and neurons [51,70,71]. This system has received considerable research attention related to its role in solid tumor cancers and possible therapies [72-74]. As the name implies HGF was originally isolated from the liver and shown to promote liver regeneration [75]. The c-Met receptor is made up of disulfide bond-linked alpha (45 kDa) and beta (145 kDa) subunits (Figure 2) [76]. c-Met’s molecular weight agrees with our estimated weight for the AT4 receptor calculated some years ago and suggests that they are the same protein. The alpha-chain of c-Metis extracellular while the beta-chain is transmembrane. HGF dimerization precedes binding to the c-Met receptor which then undergoes phosphorylation. Once phosphorylated the tyrosine residues of the beta subunit serve as docking sites for downstream signaling mediators including extracellular signal-regulated kinase (ERK) and the phosphatidylinositol-3-kinase (P13K) pathway [75].
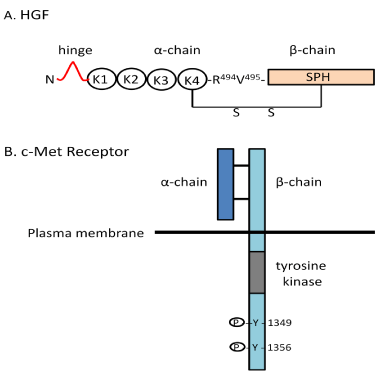
Figure 2: The structures and functions of the HGF/c-Met receptor system. (A)
The structure of hepatocyte growth factor (HGF) includes a α-chain (69 kDa) of
four Kringle domains and a β-chain (43 kDa) consisting of a serine proteinase
homology (SPH) domain linked by disulfide bonds. (B) The c-Met receptor
consists of a α-chain (50 kDa) and a β-chain (140 kDa) linked by disulfide
bonds. HGF binds to c-Met resulting in tyrosine phosphorylation leading to
a number of biological activities as listed. Nle¹-AngIV and Dihexa act at the
“hinge” region of HGF to facilitate dimerization which is a prerequisite to c-Met
receptor binding and activation.
The hypothesis that brain AngIV interacts with and activates the HGF/c-Met system is supported by the observation that AngIVassociated behavioral and physiological functions closely match those mediated by HGF [77-79]. These functions include augmentation of dendritic arborization and synaptogenesis, neurogenesis, facilitation of hippocampal long-term potentiation (LTP) and calcium signaling (hypothesized building blocks of memory consolidation), angiogenesis and facilitation of cerebral blood flow and cerebro protection. Such overlapping functions suggest that AngIV-induced activities are via activation of the HGF/c-Met system.
Recently we reported that the AT4 receptor antagonist Norleual- AngIV (Nle-YI-ψ-(CH2-NH2)-HPF) inhibited HGF binding to c-Met and in turn HGF-dependent signaling, proliferation, invasion, and scattering [78]. Norleual-AngIV’s mechanism of action as a c-Met receptor antagonist is by inhibiting the dimerization of HGF, a necessary prerequisite for binding and activation of the c-Met receptor. This dimerization process is dependent upon a short HGF domain located between its N-terminal and first kringle domain referred to as the “hinge region” (Figure 2) [80]. The importance of this hinge region was confirmed by the synthesis and utilization of a hexapeptide mimic (Hinge: KDYIRN) that bound to HGF with high affinity and blocked HGF dimerization [29]. The application of Hinge did not interfere with memory in normal functioning animals [81], a finding consistent with an earlier report that noted no impact on learning and memory in cognitively intact animals treated with AngIV and AngIV analogs [44]. Given these results we hypothesized that AngIV analogs mimic this hinge region and behave as allosteric activators by emulating the change in HGF’s conformation that normally results from its dimerization. Imaging data from cultured neonatal rat hippocampal neurons indicate that Nle¹-AngIV stimulates dendritic spine numbers and size, as well as overall dendritic arborization, suggesting a plausible mechanism for enhanced synaptic plasticity, connectivity among neurons, and facilitation of memory [28]. HGF activation of the c-Met receptor has also been shown to mediate dendritic arborization and neurogenesis in cultured hippocampal neurons [82] and facilitate memory consolidation and retrieval in memory compromised animal models [83-86]. Elevated CNS levels of HGF have been measured in patients diagnosed with multiple sclerosis, Parkinson’s disease, amyotrophic lateral sclerosis and spinal cord injury [87-90]. Unfortunately these increases in HGF are not maintained with disease progression, and the hippocampal HGF/c-Met system appears to be down regulated in AD patients [91]. Thus, the brain HGF/c-Met system appears to initially respond to neurodegenerative disease-induced injury by facilitating synaptic plasticity and neurogenesis; however these elevations in HGF are not sustained as the disease progresses.
Development of AngIV-based analogs
Given the above research findings regarding Nle¹-AngIV’s ability to facilitate synaptogenesis and memory consolidation via stimulating the HGF/c-Met system in animal models of AD AngIVbased pharmaceuticals have been proposed as therapeutic agents to treat AD (reviewed in [45,92]). In an effort to develop such a drug members of our laboratory synthesized a number of AngIV-based compounds possessing extended half-lives by utilizing reduced peptide bonds (CH2-NH2) between residues. However, two critical physiochemical properties hindered drug development: 1) a lack of metabolic stability resulting in short circulating half-lives (eg. Nle¹- AngIV = 1.42 min.; and 2) an inability to penetrate the blood-brain barrier (BBB) [93]. This latter limitation of AngIV-related peptides results from considerations of molecular size, overall hydrophobicity, and hydrogen-bonding potential as reflected by the size of the encompassing hydration sphere. We next determined that the memory facilitating effects of intra cerebroventricularly delivered Nle¹-AngIV derived from its N-terminal region given that N-directed fragments as small as tetra- and tripeptides retained the ability to overcome scopolamine-induced amnesia [2,28]. Further, Nle¹-AngIV and these shorter fragments augmented hippocampal synaptic connectivity via the formation of new synapses. Functionality of these synapses was confirmed by the presence of analog-induced spino genesis and colocalization of synaptic markers in newly formed dendritic spines, coupled with the recording of enhanced miniature excitatory postsynaptic currents. These results encouraged the possibility that a clinically useful drug could be designed possessing oral efficacy, increased metabolic stability, and BBB penetrability offering facilitated cognitive functioning. Subsequent efforts yielded the parent compound Dihexa and a related family of molecules possessing increased hydrophobicity, decreased hydrogen bonding potential, and increased metabolic stability (plasma half-life = 335 min [93]). Dihexa and its analogs bind with high affinity to HGF, induce c-Met phosphorylation in the presence of subthreshold levels of HGF, stimulate hippocampal spinogenesis and synaptogenesis equivalent with HGF [81], and promote neurogenesis and cerebro protection (data in preparation for publication). Intact Dihexa has been retrieved in cerebrospinal fluid samples taken from rats following both oral and parenteral treatment (data in preparation for publication). Treatment with the HGF antagonist, Hinge as well as a short hairpin RNA directed at c-Met, significantly inhibited these processes. These compounds penetrate the BBB in sufficient quantity to facilitate memory consolidation and retrieval in aged rats, and the scopolamine-induced amnesic rat model of AD, as measured employing the Morris water maze task of spatial memory [93].
Therapeutic prospective and limitations
Limiting side effects is of particular importance regarding angiotensin-based antihypertensive drugs given the documented problems of dry mouth, nausea and dizziness, muscle soreness, and diuresis that may occur with ACE inhibitors and ARBs (Figure 1). Each member of these classes of drugs is designed to reduce AT1 receptor activation and control hypertension. However, the AngII/AT1 receptor system influences multiple functions beyond blood pressure, including body water balance, control of vasopressin and oxytocin release and sexual reproduction and behavior, thus undesirable drug-induced effects are possible. Dihexa-based compounds do not interact with central or peripheral AT1 receptors, are highly target specific, exhibit little interaction with cardiac channel proteins and hepatic CIP isoforms, and reveal no acute toxicity following a 6x effective dose of Dihexa. More extensive safety studies are currently underway. These data predict that the greatest clinical impact will be in individuals with compromised brain HGF/c-Met systems as present in early-stage AD. The combined neuroprotective, synaptogenic, and neurogenic mechanisms proposed for these compounds encourage the possibility that they may be a treatment option for neurodegenerative and neuro-traumatic disorders beyond AD. Despite the potential to attenuate and possibly reverse deleterious molecular events common to many neurodegenerative diseases, we do not foresee this approach as a “cure” because the underlying etiologies will likely persist. We do believe that damage due to ongoing neurodegenerative processes will be significantly slowed and attenuated.
Conclusion
New pharmacological approaches to treat AD include anti- amyloid and anti-Tau drugs to clear cellular Aβ and Tau proteins respectively, NSAIDs, selective COX-2 inhibitors, Gamma-secretase modulators, and anti-amyloid antibodies to block β-amyloid storage. While harnessing the regenerative capacity of neurotrophic factors has been considered as a treatment approach to dementia practical implementation of this concept has been lacking. Recently, HGFdirected molecules have been synthesized that are orally active and possess sufficient BBB permeability to facilitate improved cognitive function in animal models of AD. The therapeutic value of this approach lies in its capacity to encourage the formation of new functional synaptic connections among existing neurons, and facilitate the replacement of damaged and lost neurons from available neural stem cell populations. These treatment outcomes would benefit patients afflicted with AD and perhaps individuals with other neurodegenerative diseases.
Acknowledgement
Drs. Wright and Harding are co-founders of M3 Biotechnology, Inc. and hold stock in this company which is involved in the development of AD and Parkinson’s disease drugs. Dr. Kawas is the CEO of M3. No funds from M3 were used in the writing of this manuscript. Although the authors have financial interest in M3 we acknowledge no conflict of interest in the preparation of this review article which represents an impartial and accurate presentation of research findings. All animal experiments conducted in our laboratory and presented in this manuscript adhered to the Guidelines for the Care and Use of Laboratory Animals as required by the National Institutes of Health (NIH Publication No. 80-23), and these protocols were approved by the Washington State University Institutional Animal Care and Use Committee.
References
- Bloom GS. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014; 71: 505-508.
- Wright JW, Harding JW. The brain angiotensin IV/AT4 receptor system as a new target for the treatment of Alzheimer’s disease. Drug Dev Res. 2009; 70: 472-480.
- Honig LS, Boyd CD. Treatment of Alzheimer's Disease: Current Management and Experimental Therapeutics. Curr Transl Geriatr Exp Gerontol Rep. 2013; 2: 174-181.
- Hurd MD, Martorell P, Delavande A, Mullen KJ, Langa KM. Monetary costs of dementia in the United States. N Engl J Med. 2013; 368: 1326-1334.
- Parsons CG, Danysz W, Dekundy A, Pulte I. Memantine and cholinesterase inhibitors: complementary mechanisms in the treatment of Alzheimer's disease. Neurotox Res. 2013; 24: 358-369.
- Wilkinson D. A review of the effects of memantine on clinical progression in Alzheimer's disease. Int J Geriatr Psychiatry. 2012; 27: 769-776.
- Suehs BT, Davis CD, Alvir J, Van Amerongen D, Pharmd NC, Joshi AV, et al. The clinical and economic burden of newly diagnosed Alzheimer’s disease in a medicare advantage population. Am J Alzheimers Dis Other Dement. 2013; 28: 384-392.
- Brayne C, Matthews FE, Xuereb JH. Pathological correlates of late onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet. 2001; 357: 169-175.
- Chen M, Maleski JJ, Sawmiller DR. Scientific truth or false hope? Understanding Alzheimer's disease from an aging perspective. J Alzheimers Dis. 2011; 24: 3-10.
- Ding Q, Dimayuga E, Keller JN. Proteasome regulation of oxidative stress in aging and age-related diseases of the CNS. Antioxid Redox Signal. 2006; 8: 163-172.
- Polidori MC, Pientka L. Bridging the pathophysiology of Alzheimer's disease with vascular pathology: the feed-back, the feed-forward, and oxidative stress. J Alzheimers Dis. 2012; 28: 1-9.
- de la Torre JC. Carotid artery ultrasound and echocardiography testing to lower the prevalence of Alzheimer's disease. J Stroke Cerebrovasc Dis. 2009; 18: 319-328.
- Cohen AD, Klunk WE. Early detection of Alzheimer's disease using PiB and FDG PET. Neurobiol Dis. 2014; 72 Pt A: 117-122.
- Mormino EC, Betensky RA, Hedden T, Schultz AP, Amariglio RE, Rentz DM, et al. Synergistic effect of ß-amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurol. 2014; 71: 1379-1385.
- Albiston AL, Diwakarla S, Fernando RN, Mountford SJ, Yeatman HR, Morgan B, et al. Identification and development of specific inhibitors for insulin-regulated aminopeptidase as a new class of cognitive enhancers. Br J Pharmacol. 2011; 164: 37-47.
- Melnikova I. Therapies for Alzheimer's disease. Nat Rev Drug Discov. 2007; 6: 341-342.
- Thomas SJ, Grossberg GT. Memantine: a review of studies into its safety and efficacy in treating Alzheimer's disease and other dementias. Clin Interv Aging. 2009; 4: 367-377.
- Cook C, Stankowski JN, Carlomagno Y, Stetler C, Petrucelli L. Acetylation: a new key to unlock tau's role in neurodegeneration. Alzheimers Res Ther. 2014; 6: 29.
- de la Torre JC, Stefano GB. Evidence that Alzheimer's disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Brain Res Rev. 2000; 34: 119-136.
- Di Marco LY, Venneri A, Farkas E, Evans PC, Marzo A, Frangi AF. Vascular dysfunction in the pathogenesis of Alzheimer's disease--A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol Dis. 2015; 82: 593-606.
- Heneka M, Andreasson K, Bachstetter AD, Colonna M, Ginhoux F, Holmes C, et al. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J Neurochem. 2016.
- Miguel-Alvarez M, Santos-Lozano A, Sanchis-Gaomar F, Fluza-Luces C, Pareja-Galeano H, Garatachea N, et al. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: A systematic review and meta-analysis of treatment effect. Drugs Aging. 2015; 32: 139-147.
- Nabavi SF, Braidy N, Gortzi O, Sobarzo-Sanchez E, Daglia M, Skalicka-Woźniak K, et al. Luteolin as an anti-inflammatory and neuroprotective agent: A brief review. Brain Res Bull. 2015; 119: 1-11.
- Sgarbossa A, Giacomazza D, di Carlo M. Ferulic Acid: A Hope for Alzheimer's Disease Therapy from Plants. Nutrients. 2015; 7: 5764-5782.
- Boyce VS, Mendell LM. Neurotrophic factors in spinal cord injury. Handb Exp Pharmacol. 2014; 220: 443-460.
- Lu B, Nagappan G, Lu Y. BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol. 2014; 220: 223-250.
- Williams AJ, Umemori H. The best-laid plans go oft awry: synaptogenic growth factor signaling in neuropsychiatric disease. Front Synaptic Neurosci. 2014; 6: 4.
- Benoist CC, Wright JW, Zhu M, Appleyard SM, Wayman GA, Harding JW. Facilitation of hippocampal synaptogenesis and spatial memory by C-terminal truncated Nle¹-angiotensin IV analogs. J Pharmacol Exp Ther. 2011; 339: 35-44.
- Kawas LH, Yamamoto BJ, Wright JW, Harding JW. Mimics of the dimerization domain of hepatocyte growth factor exhibit anti-Met and anticancer activity. J Pharmacol Exp Ther. 2011; 339: 509-518.
- Kawas LH, McCoy AT, Yamamoto BJ, Wright JW, Harding JW. Development of angiotensin IV analogs as hepatocyte growth factor/Met modifiers. J Pharmacol Exp Ther. 2012; 340: 539-548.
- Wright JW, Harding JW. Brain renin-angiotensin--a new look at an old system. Prog Neurobiol. 2011; 95: 49-67.
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010; 140: 918-934.
- Phillips MI, de Oliveira EM. Brain renin angiotensin in disease. J Mol Med (Berl). 2008; 86: 715-722.
- Di Marco LY, Venneri A, Farkas E, Evans PC, Marzo A, Frangi AF. Vascular dysfunction in the pathogenesis of Alzheimer’s disease - A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol Dis. 2015; 82: 593-606.
- Ohshima K, Mogi M, Horiuchi M. Therapeutic approach for neuronal disease by regulating renin-angiotensin system. Curr Hypertens Rev. 2013; 9: 99-107.
- Ownby RL. Neuroinflammation and cognitive aging. Curr Psychiatry Rep. 2010; 12: 39-45.
- Gard PR. Angiotensin as a target for the treatment of Alzheimer's disease, anxiety and depression. Expert Opin Ther Targets. 2004; 8: 7-14.
- Mustafa T, Lee JH, Chai SY, Albiston AL, McDowall SG, Mendelsohn FA. Bioactive angiotensin peptides: focus on angiotensin IV. J Renin Angiotensin Aldosterone Syst. 2001; 2: 205-210.
- Ongali B, Nicolakakis N, Tong XK. Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiol Dis. 2014; 68: 126-136.
- Harding JW, Cook VI, Miller-Wing AV, Hanesworth JM, Sardinia MF, Hall KL, et al. Identification of an AII(3-8) [AIV] binding site in guinea pig hippocampus. Brain Res. 1992; 583: 340-343.
- Wright JW, Harding JW. The Brain Hepatocyte Growth Factor/c-Met Receptor System: A New Target for the Treatment of Alzheimer's Disease. J Alzheimers Dis. 2015; 45: 985-1000.
- Braszko JJ, Walesiuk A, Wielgat P. Cognitive effects attributed to angiotensin II may result from its conversion to angiotensin IV. J Renin Angiotensin Aldosterone Syst. 2006; 7: 168-174.
- Nade VS, Kawale LA, Valte KD, Shendye NY. Cognitive enhancing effect of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers on learning and memory. Indian J Pharmacol. 2015; 47: 263-269.
- Wright JW, Stubley L, Pederson ES, Kramár EA, Hanesworth JM, Harding JW. Contributions of the brain angiotensin IV-AT4 receptor subtype system to spatial learning. J Neurosci. 1999; 19: 3952-3961.
- Wright JW, Kawas LH, Harding JW. The development of small molecule angiotensin IV analogs to treat Alzheimer's and Parkinson's diseases. Prog Neurobiol. 2015; 125: 26-46.
- de la Torre JC1. Alzheimer's disease: how does it start? J Alzheimers Dis. 2002; 4: 497-512.
- Chai SY, Bastias MA, Clune EF, Matsacos DJ, Mustafa T, Lee JH, et al. Distribution of angiotensin IV binding sites (AT4 receptor) in the human forebrain, midbrain and pons as visualized by in vitro receptor autoradiography. J ChemNeuroanat. 2000; 20: 339-348.
- Park H1, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013; 14: 7-23.
- Cohen S, Levi-Montalcini R, Hamburger V. A nerve growth-stimulating factor isolated from sarcom as 37 and 180. Proc Natl Acad Sci U S A. 1954; 40: 1014-1018.
- Barde YA, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982; 1: 549-553.
- Bottaro DP1, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991; 251: 802-804.
- Koike H, Ishida A, Shimamura M, Mizuno S, Nakamura T, Ogihara T, et al. Prevention of onset of Parkinson’s disease by in vivo gene transfer of human hepatocyte growth factor in rodent model: A model of gene therapy for Parkinson’s disease. Gene Ther. 2006; 13: 1639-1644.
- Martins GJ1, Plachez C, Powell EM. Loss of embryonic MET signaling alters profiles of hippocampal interneurons. Dev Neurosci. 2007; 29: 143-158.
- Takeuchi D1, Sato N, Shimamura M, Kurinami H, Takeda S, Shinohara M, et al. Alleviation of Abeta-induced cognitive impairment by ultrasound-mediated gene transfer of HGF in a mouse model. Gene Ther. 2008; 15: 561-571.
- Cho T, Ryu JK, Taghibiglou C, Ge Y, Chan AW, Liu L, et al. Long-term potentiation promotes proliferation/survival and neuronal differentiation of neural stem/progenitor cells. PloS One. 2013; 10: e76860.
- Fitzsimons CP, van Bodegraven E, Schouten M, Lardenoije R, Kompotis K, Kenis G, et al. Epigenetic regulation of adult neural stem cells: implications for Alzheimer's disease. Mol Neurodegener. 2014; 9: 25.
- Woo KW, Kwon OW, Kim SY, Choi SZ, Son MW, Kim KH, Lee KR. Phenolic derivatives from the rhizomes of Dioscoreanipponica and their anti-neuroinflammatory and neuroprotective activities. J Ethnopharmacol. 2014; 155: 1164-1170.
- Forlenza OF, Diniz BS, Teixeira AL, Radanovic M, Talib LL, Racha NP, et al. Lower cerebrospinal fluid concentration of brain-derived neurotrophic factor predicts progression from mild cognitive impairment to Alzheimer’s disease. Neuromolecular Med. 2015; 17: 326-332.
- Lu B, Nagappan G, Guan X, Nathan PH, Wren P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci. 2013; 14: 401-416.
- Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011; 10: 209-219.
- Song JH, Yu JT, Tan L. Brain-Derived Neurotrophic Factor in Alzheimer's Disease: Risk, Mechanisms, and Therapy. Mol Neurobiol. 2015; 52: 1477-1493.
- Adachi N, Numakawa T, Richards M, Richards M, Nakajima S, Kunugi H. New insight in expression, transport, and secretion of brain-derived neurotrophic factor: Implications in brain-related diseases. World J Biol Chem. 2014; 5: 409-428.
- Zhang L, Fang Y, Lian Y, Chen Y, Wu T, Zheng Y, et al. Brain-derived neurotrophic factor ameliorates learning deficits in a rat model of Alzheimer’s disease induced by aß1-42. Plos One. 2015; 10: e0122415.
- Koo JH, Kwon IS, Kang EB, Lee CK, Lee NH3, Kwon MG, et al. Neuroprotective effects of treadmill exercise on BDNF and PI3-K/Akt signaling pathway in the cortex of transgenic mice model of Alzheimer's disease. J Exerc Nutrition Biochem. 2013; 17: 151-160.
- Aloe L, Rocco ML. NGF and therapeutic prospective: what have we learned from the NGF transgenic models? Ann Ist Super Sanita. 2015; 51: 5-10.
- Tuszynski MH, Yang JH, Barba D, U HS, Bakay RA, Pay MM, et al. Nerve growth factor gene therapy: Activation of neuronal responses in Alzheimer Disease. JAMA Neurol. 2015; 72:1139-1147.
- Bothwell M. NGF, BDNF, NT3, and NT4. Handb Exp Pharmacol. 2014; 220: 3-15.
- Gómez-Palacio-Schjetnan A, Escobar ML. Neurotrophins and synaptic plasticity. Curr Top Behav Neurosci. 2013; 15: 117-136.
- Schindowski K, Belarbi K, Buée L. Neurotrophic factors in Alzheimer's disease: role of axonal transport. Genes Brain Behav. 2008; 1: 43-56.
- Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003; 22: 309-325.
- Skead G, Govender D. Gene of the month: MET. J Clin Pathol. 2015; 68: 405-409.
- Awad AJ, Burns TC, Zhang Y, Abounader R. Targeting MET for glioma therapy. Neurosurg Focus. 2014; 37: E10.
- Benvenuti S, Comoglio PM. The MET receptor tyrosine kinase in invasion and metastasis. J Cell Physiol. 2007; 213: 316-325.
- Jiang WG, Martin TA, Parr C, Davies G, Matsumoto K, Nakamura T. Hepatocyte growth factor, its receptor, and their potential value in cancer therapies. Crit Rev Oncol Hematol. 2005; 53: 35-69.
- Nakamura T, Mizuno S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc Jpn Acad Ser B Phys Biol Sci. 2010; 86: 588-610.
- Shinomiya N, Vande Woude GF. Suppression of met expression: a possible cancer treatment. Commentary re: S. J. Kim et al., reduced c-Met expression by an adenovirus expressing a c-Met ribozyme inhibits tumorigenic growth and lymph node metastases of PC3-LN4 prostate tumor cells in an orthotopic nude mouse model. Clin. Cancer Res., 14: 5161-5170, 2003. Clin Cancer Res. 2003; 9: 5085-5090.
- Qiu S, Lu Z, Levitt P. MET receptor tyrosine kinase controls dendritic complexity, spine morphogenesis, and glutamatergic synapse maturation in the hippocampus. J Neurosci. 2014; 34: 16166-16179.
- Yamamoto BJ, Elias PD, Masino JA, Hudson BP, McCoy AT, Anderson ZJ, et al. The angiotensin IV analog Nle-Tyr-Leu-?-(CH2-NH2)3-4-His-Pro-Phe (Norleual) can act as a hepatocyte growth factor/c-Met inhibitor. J Pharmacol Exp Ther. 2010; 333: 161-173.
- Zeng W, Ju R, Mao M. Therapeutic potential of hepatocyte growth factor against cerebral ischemia (Review). ExpTher Med. 2015; 9: 283-288.
- Youles M, Holmes O, Petoukhov MV, Nessen MA, Stivala S, Svergan DI, et al. Engineering the NK1 fragment of hepatocyte growth factor/scatter factor as a MET receptor antagonist. J Mol Biol. 2008; 377: 616-622.
- Benoist CC, Kawas LH, Zhu M, Tyson KA, Stillmaker L, Appleyard SM. The procognitive and synaptogenic effects of angiotensin IV-derived peptides are dependent on activation of the hepatocyte growth factor/c-met system. J Pharmacol Exp Ther. 2014; 351: 390-402.
- Tyndall SJ, Walikonis RS. Signaling by hepatocyte growth factor in neurons is induced by pharmacological stimulation of synaptic activity. Synapse. 2007; 61: 199-204.
- Date I, Takagi N, Takagi K, Kago T, Matsumoto K, Nakamura T, et al. Hepatocyte growth factor attenuates cerebral ischemia-induced learning dysfunction. Biochem Biophysics Res Comm. 2004; 319: 1152-1158.
- Date I, Takagi N, Takagi K, Kago T, Matsumoto K, Nakamura T. Hepatocyte growth factor improved learning and memory dysfunction of microsphere-embolized rats. J Neurosci Res. 2004; 78: 442-453.
- Kato T, Funakoshi H, Kadoyama K, Noma S, Kanai M, Ohya-Shimada W, et al. Hepatocyte growth factor overexpression in the nervous system enhances learning and memory performance in mice. J Neurosci Res. 2012; 90: 1743-1755.
- Shimamura M, Sato N, Waguri S, Uchiyama Y, Hayashi T, Iida H, et al. Gene transfer of hepatocyte growth factor gene improves learning and memory in the chronic stage of cerebral infarction. Hypertension. 2006; 47: 742-751.
- Kato S, Funakoshi H, Nakamura T, Noma S, Kanai M, Ohya-Shimada W, et al. Expression of hepatocyte growth factor and c-Met in the anterior horn cells of the spinal cord in the patients with amyotrophic lateral sclerosis (ALS): Immunohistochemical studies on sporadic ALS and familial ALS with superoxide dismutase 1 gene mutation. Acta Neurophathol. 2003; 106: 112-120.
- Muller AM, Jun E, Conlon H, Sadiq SA. Cerebrospinal hepatocyte growth factor levels correlate negatively with disease activity in multiple sclerosis. J Neuroimmunol. 2012; 251: 80-86.
- Salehi Z, Rajaei F. Expression of hepatocyte growth factor in the serum and cerebrospinal fluid of patients with Parkinson's disease. J Clin Neurosci. 2010; 17: 1553-1556.
- Shimamura M, Sato N, Sata M, Wakayama K, Ogihara T, Morishita R. Expression of hepatocyte growth factor and c-Met after spinal cord injury in rats. Brain Res. 2007; 1151: 188-194.
- Hamasaki H, Honda H, Suzuki SO, Hokama M, Kiyohara Y, Nakabeppu Y, et al. Down-regulation of MET in hippocampal neurons of Alzheimer's disease brains. Neuropathology. 2014; 34: 284-290.
- Wright JW, Kawas LH, Harding JW. A Role for the Brain RAS in Alzheimer's and Parkinson's Diseases. Front Endocrinol (Lausanne). 2013; 4: 158.
- McCoy AT, Benoist CC, Wright JW, Kawas LH, Bule-Ghogare JM, Zhu M, et al. Evaluation of metabolically stabilized angiotensin IV analogs as procognitive/antidementia agents. J Pharmacol Exp Ther. 2013; 344: 141-154.