
Mini Review
Austin J Clin Neurol 2016;3(2): 1091.
Cholinesterase Inhibitors as a Disease-Modifying Therapy for Alzheimer’s Disease: The Anticholinergic Hypothesis
Hori K1,2*, Hosoi M², Konishi K³, Sodenaga M¹, Hashimoto C¹, Sasaki O¹, Suzuki I¹, Maedomari M¹, Tadokoro M¹, Tsukahara S¹, Kamatani H4, Kocha H¹, Tomioka H2 and Hachisu M5
¹Department of Neuropsychiatry, St. Marianna University, Japan
²Department of Psychiatry, Showa University Northern Yokohama Hospital, Japan
³Tokyo Metropolitan Tobu Medical Center for Persons with Developmental/ Multiple Disabilities, Japan
4Department of Psychiatry, Kawasaki Memorial Hospital, Japan
5Department of Pharmaceutical Therapeutics, Division of Clinical Pharmacy, Showa University, Japan
*Corresponding author: Koji Hori, Department of Neuropsychiatry, St. Marianna University School of Medicine, 2-16-1 Sugao, Miyamaeku, Kawasaki City, Kanagawa, Japan
Received: August 08, 2016; Accepted: September 20, 2016; Published: September 23, 2016
Abstract
In this mini-review article, we summarize our previous results and discuss whether cholinesterase inhibitors (ChEIs) should be considered a diseasemodifying therapy for Alzheimer’s disease (AD). First, we reiterate that the acceleration in AD progression is related to the endogenous appearance of anticholinergic activity (AA) and that one of the two possible pathways generating amyloid peptides in AD is related to acetylcholine downregulation. Second, we compare the kinetics of ACh between the mild cognitive impairment stage and normal stage according to the hypothesis of AA in AD. Third, we differentiate among ChEIs according to the classification of AD stage based on AA and also speculate that ChEIs might be useful as disease-modifying therapies.
Keywords: Anticholinergic activity; Acetylcholine; Alzheimer’s disease; Choline acetyltransferase; Disease-modifying therapy; Mild cognitive impairment; Pharmacotherapy
Abbreviations
AA: Anticholinergic Activity; ACh: Acetylcholine; AD: Alzheimer’s Disease; BuChE: Butyrylcholinesterase; ChAT: Choline Acetyltransferase; ChEI: Cholinesterase Inhibitor; MCI: Mild Cognitive Impairment; NMDA: N-methyl-D-aspartate
Introduction
Antidementia agents such as cholinesterase inhibitors (ChEIs) and N-methyl-D-aspartate (NMDA) receptor antagonists are not considered disease-modifying therapies but symptomatic treatments. However, based on our theory of the endogenous appearance of anticholinergic activity (AA) in Alzheimer’s disease (AD) [1,2], we believe that ChEIs and NMDA receptor antagonists should be considered both disease-modifying therapies and symptomatic treatments [3].
Therefore, in this mini-review article, we discuss why ChEIs should be considered a disease-modifying therapy based on our previous work. First, we summarize why the acceleration in AD progression is believed to be related to the endogenous appearance of AA in AD [1] and why one of the two pathways generating amyloid peptides in AD is related to ACh downregulation [2]. Second, we reiterate our hypothesis that the mild cognitive impairment (MCI) and mild stages of AD can be explained by the hypothesis of endogenous AA in AD [1,2] and hypothesize that the kinetics of ACh are altered in MCI stages compared with the normal stage. Third, we differentiate among ChEIs according to the classification of AD stage based on AA levels and speculate that ChEIs are disease-modifying therapies.
Classification of AD based on the hypothesis of endogenous AA in AD
ACh is related not only to cognitive function but also regulation of inflammation [4-7]. AD patients show ACh downregulation [8,9], which might possibly up regulate the inflammation found in the brains of AD patients. This inflammation might induce the release of cytokines with AA. Therefore, we previously hypothesized that AA both in the central nervous system and peripheral tissue might appear endogenously in the moderate stage of AD [1,2]. At this stage, the depression of the cholinergic system reaches a critical level, with increased NMDA receptor expression causing hyperactivity of the inflammatory system and leading to AA. Accordingly, we proposed our hypothesis of endogenous AA in AD [1,2]. AA is considered not only to depress ACh by antagonizing its binding to the ACh receptor but also to induce amyloid production [10,11]. Thus, we speculated that AD progresses more rapidly at the moderate stage than at the mild stage due to endogenous AA [12].
We therefore believe that there are two amyloidogenic patterns in AD. The first pattern (P1 pattern) is pathological and is unrelated to the ACh downregulation observed in MCI or mild AD. The second pattern (P2 pattern) is also pathological but is related to the ACh downregulation observed in moderate AD. The P1 pattern probably begins when the AD pathology occurs and is likely to be misdiagnosed as normal aging due to the slow decline in cognitive function. However, the P2 pattern is related to ACh downregulation and is clearly prominent, and it can be readily diagnosed as AD at the moderate stage when AD patients present with clinical symptoms such as memory disturbance, disorientation, aphasia, delusions, hallucinations, and diurnal rhythm disturbance [3,13]. The decline is also more rapid at the moderate stage than in patients with MCI or mild AD [12]. Therapeutically, we recommend that ChEIs and NMDA receptor antagonists be prescribed for the prevention and treatment of the appearance of AA and the rapid progression of AD, respectively [3]. Thus, ChEIs and NMDA receptor antagonists should be considered both disease-modifying therapies and symptomatic treatments [3]. As for NMDA receptor antagonists, these medications abolish AA due to hyperactivation of ACh and slow the progression of AD (Figure 1) [13,14].
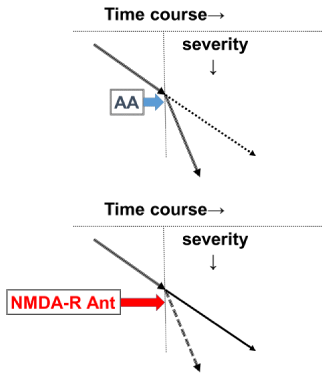
Figure 1: In moderate-stage AD, AA appears endogenously and the
progression of AD is accelerated (upper panel). This process involves NMDA
receptor hyperactivity. Therefore, NMDA-R Ant can abolish AA and reduce
the speed of AD progression to that of the mild stage (lower panel). This
figure is reproduced from Hori, et al. [13]. AA: Anticholinergic Activity; AD:
Alzheimer’s Disease; NMDA: N-methyl-D-aspartate; NMDA-R Ant: NMDA
Receptor Antagonist.
Moreover, we also speculate that there might be a compensatory mechanism in relatively early-stage AD. Generally speaking, AD pathology causes the degeneration of cholinergic neurons [6,7]. However, although the activity of choline acetyltransferase (ChAT, the enzyme that produces ACh) is down regulated in the mild and moderate stages of AD, increased ChAT activity has also been reported in patients with MCI or early AD [15-17] and in the tau rat model [18]. Hara, et al. [18] reported that acetylcholinesterase upregulation in the septum may result in the selective degeneration of the septohippocampal cholinergic pathway in the tauopathy mouse model. Our group and other group also previously speculated that this upregulation of ChAT is related to a compensatory mechanism [15-17,19]. This mechanism ensures that the ACh level is relatively normal and that cognitive function remains largely intact. This compensatory reaction to the onset of AD may be attributable to hyperactive presynaptic cholinergic neurons. If this compensatory mechanism works, then the cholinergic system remains intact, rather than deteriorated. Thus, when clinical symptoms occur, the cholinergic system is burdened but functioning and the neurons are functional (i.e., not degenerated).
Based on these speculations, we hypothesize that, in the MCI stage, the AD pathology burdens the brains of AD patients, but that ChAT activity is up regulated, resulting in normal ACh levels. Moreover, we propose that ACh gradually decreases in mild-stage AD. The hyperactivity of presynaptic neurons may cause early and rapid neuronal degeneration and depress ChAT activity [15-17]. Therefore, we consider it important to maintain the compensatory reaction as long as possible. To do this, it is important to down regulate cholinesterase so as not to up regulate ChAT activity and hyper activate ACh (Figure 2) [19].
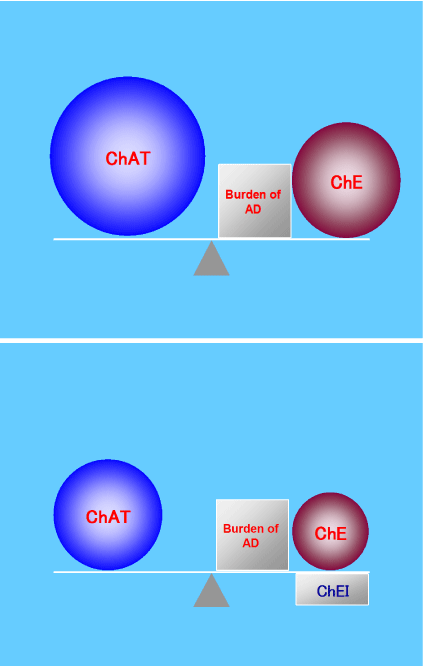
Figure 2: In the MCI stage, ChAT is hyper activated to compensate for
the AD burden and to maintain ACh at a normal level. However, this ChAT
upregulation causes early degeneration of ACh neurons (upper panel).
Therefore, it is important to down regulate ChE activity to maintain a normal
ACh level without ChAT upregulation. For this reason, we should prescribe
ChEIs to patients with AD, even those with MCI (lower panel). These figures
are reproduced from Konishi, et al. [19], with the permission of Karger, Basel,
Switzerland.
ACh: Acetylcholine; AD: Alzheimer’s Disease; ChAT: Choline
Acetyltransferase; ChE: Cholinesterase; ChEI: Cholinesterase Inhibitor;
MCI: Mild Cognitive Impairment.
ACh kinetics in the MCI and normal stages and the differences among ChEIs
The compensatory mechanism might succeed in the MCI stage and support the cholinergic system. However, there might be two situations in which ACh is down regulated or overburdened in MCI that are not seen in the normal stage. One occurs when patients with AD at the MCI stage relax, at which time the ACh level is lower than normal. In this situation, these patients show apathy (e.g., when they watch television, they fall asleep). The other situation is when they are more stressed than usual. In this situation, their cholinergic system cannot be up regulated any further because ChAT is already activated and does not permit further upregulation (Figure 2). We have discussed this mechanism above. When the ACh system is intact and not depressed or overloaded, upregulation of ACh is possible, even in the presence of another AA-inducing factor (e.g., medication [20], febrile illness [21], or mental stress [22]. Consequently, the inflammatory system is not up regulated and AA does not appear. Even if ACh downregulation does not reach a critical level, when another AA-inducing factor is present and the ACh system is depressed or overloaded, the inflammatory system is up regulated and AA appears [23].
Alternatively, when the ACh system is intact and not depressed or overloaded, it can compensate for the effects of other AA inserts and stress is ameliorated. However, when the ACh system is overloaded, such as in the MCI stage, it cannot compensate for other AA inserts and the stress continues, causing depression. In this situation, ChEI prescription may enable ACh upregulation by boosting ChAT (Figure 3). The other implication of this hypothesis is that patients who show apathy when relaxed or who show depressive symptoms or irritability related to depressive symptoms in AD could be diagnosed with AD (at the MCI stage) even when they do not show cognitive impairment, permitting early diagnosis of AD. Alternatively, the cognitive functions of patients with MCI are largely maintained. Therefore, in the clinical setting, patients get relatively high scores on the mental examination test battery. We would also be able to predict the speed of cognitive decline in these patients. If the patients show apathy, depressive symptoms, or irritability despite good cognitive function, we can speculate that the cognitive decline in these patients would be faster than that of normal aging and thus diagnose them with AD (Figure 4).
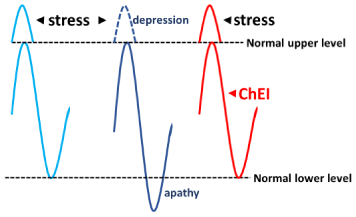
Figure 3: There might be two situations in which ACh is down regulated or
overburdened in MCI (middle) that are not seen in the normal stage (left). One
occurs when patients with AD at the MCI stage relax, at which time the ACh
level is lower than normal. In this situation, these patients show apathy (e.g.,
when they watch television, they fall asleep). The other situation is when they
are more stressed than usual. In this situation, their cholinergic system cannot
be up regulated any further because ChAT is already activated and does not
permit further upregulation. When ChEIs are prescribed, ACh upregulation
is possible, even if another AA-inducing factor is present, to compensate for
other AA inserts and ameliorate stress (right).
AA: anticholinergic activity; ACh: Acetylcholine; AD: Alzheimer’s Disease;
ChAT: Choline Acetyltransferase; ChEI: Cholinesterase Inhibitor; MCI: Mild
Cognitive Impairment.
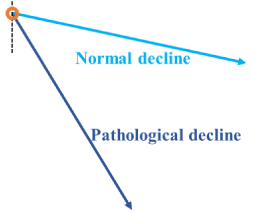
Figure 4: Cognitive functions in patients with MCI are relatively maintained.
Therefore, in the clinical setting, patients obtain relatively high scores on
the mental examination test battery. It is important to predict the speed of
decline in these patients (orange circle). Patients showing apathy, depressive
symptoms, or irritability even with good cognitive function will probably have a
faster rate of decline (pathological decline; dark blue line) than during normal
aging (normal decline; light blue line) and be diagnosed with AD. When
patients show apathy when relaxed or depressive symptoms or irritability
that is related to depressive symptoms in AD, they should be diagnosed with
AD (at the MCI stage) even when they do not show cognitive impairment,
permitting the early diagnosis of AD.
AD: Alzheimer’s Disease; MCI: Mild Cognitive Impairment.
Finally, we should differentiate among the three ChEIs we prescribe. Because the cholinergic system is considered to be intact in the MCI stage, nicotinic receptor, amyloid, glia, and butyrylcholinesterase (BuChE) levels are also considered to be normal. Therefore, donepezil should be prescribed because it specifically inhibits acetylcholinesterase. In contrast, in mild-stage disease, particularly in the mild and moderate stages, we should consider prescribing rivastigmine or galantamine because this stage shows a gradual decrease in ACh that depresses ChAT activity due to cholinergic neuron degeneration and nicotinic receptor downregulation. Of course, BuChE is also up regulated because of the proliferation of amyloid peptides and glia cells [24]. When patients show marked apathy, we prescribe rivastigmine. Moreover, rivastigmine should be prescribed to younger patients because amyloid pathology is more predominant than the physiological aging process in these patients compared with older AD patients [25]. In contrast, when patients show marked depressive symptoms or irritability, we should prescribe galantamine [26].
Based on these hypotheses, we consider ChEIs to be not only a symptomatic treatment but also a disease-modifying therapy (Figure 5). NMDA receptor antagonists also have disease-modifying properties. It is vital to develop new pharmaceutical approaches to AD. However, we also consider it important to reevaluate already available medicines, namely, ChEIs and NMDA receptor antagonists, as disease-modifying therapies.
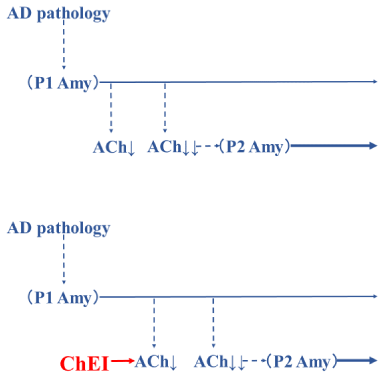
Figure 5: There may be two steps by which amyloid is generated. The
first pattern (P1 pattern) is caused by AD pathology and induces ACh
downregulation. The second pattern is another pathological pattern (P2
pattern) and is related to the ACh downregulation. When the downregulation of
ACh caused by the P1 amyloid pattern reaches a critical level, the P2 amyloid
pattern develops, accelerating the progression of AD (upper). If ChEIs are
prescribed, the time to the critical ACh downregulation and the initiation of the
P2 amyloid pattern is delayed. Therefore, ChEI is a symptomatic treatment
for the P1 amyloid pattern and is also a disease-modifying treatment of P2
amyloid (lower). These figures were produced based on Hori, et al. [3].
ACh: Acetylcholine; AD: Alzheimer’s Disease; Amy: Amyloid; ChEI:
Cholinesterase Inhibitor.
References
- Hori K, Konishi K, Akita R, Tani M, Tomioka H, Kitajima Y, et al. [Proposal of endogenous anticholinergic hypothesis in Alzheimer disease]. Nihon Shinkei Seishin Yakurigaku Zasshi. 2013; 33: 117-126.
- Hori K, Konishi K, Tani M, Tomioka H, Akita R, Kitajima Y, et al. Serum anticholinergic activity: a possible peripheral marker of the anticholinergic burden in the central nervous system in Alzheimer’s disease. Dis Markers. 2014; 2014: 459013.
- Hori K, Konishi K, Tani M, Tomioka H, Akita R, Kitajima Y, et al. Why does the progression of Alzheimer’s disease accelerate? Ann Psychiatry Ment Health. 2014; 2: 1006.
- Pavlov VA, Ochani M, Gallowitsch-Puerta M, Ochani K, Huston JM, Czura CJ, et al. Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc Natl Acad Sci USA. 2006; 103: 5219- 5223.
- Mabley JG, Pacher P, Szabo C. Activation of the cholinergic antiinflammatory pathway reduces ricin-induced mortality and organ failure in mice. Mol Med. 2009; 15: 166-172.
- Lykhmus O, Koval L, Pastuhova D, Zouridakis M, Tzartos S, Komisarenko S, et al. The role of carbohydrate component of recombinant a7 nicotinic acetylcholine receptor extracellular domain in its immunogenicity and functional effects of resulting antibodies. Immunobiology. 2016; S0171-S2985.
- Di Bari M, Di Pinto G, Reale M, Mengod G, Tata AM. Cholinergic system and neuroinflammation: Implication in multiple sclerosis. Cent Nerv Syst Agents Med Chem. 2016.
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982; 215: 1237-1239.
- Gil-Bea FJ, Solas M, Mateos L, Winblad B, Ramírez MJ, Cedazo-Mínguez A. Cholinergic hypofunction impairs memory acquisition possibly through hippocampal Arc and BDNF downregulation. Hippocampus. 2011; 21: 999- 1009.
- Perry EK, Kilford L, Lees AJ, Burn DJ, Perry RH. Increased Alzheimer pathology in Parkinson’s disease related to antimuscarinic drugs. Ann Neurol. 2003; 54: 235-238.
- Lu CJ, Tune LE. Chronic exposure to anticholinergic medications adversely affects the course of Alzheimer disease. Am J Geriatr Psychiatry. 2003; 11: 458-461.
- Konishi K, Hori K, Tani M, Tomioka H, Kitajima Y, Akashi N, et al. Hypothesis of Endogenous Anticholinergic Activity in Alzheimer’s Disease. Neurodegener Dis. 2015; 15: 149-156.
- Hori K, Konishi K, Hachisu M. Serum anticholinergic activity: relationship with clinical symptoms in Alzheimer’s disease and proposal of new biological marker. Nihon Shinkei Seishin Yakurigaku Zasshi. 2011; 31: 135-140.
- Hori K, Konishi K, Minegishi G, Tomioka H, Tani M, Tanaka H, et al. Memantine abolishes anticholinergic activity in patient with Alzheimer’s disease at moderate stage. J Alzheimer Dis Parkinsonism. 2012; 2: 108.
- Gilmor ML, Erickson JD, Varoqui H, Hersh LB, Bennett DA, Cochran EJ, et al. Preservation of nucleus basalis neurons containing choline acetyltransferase and the vesicular acetylcholine transporter in the elderly with mild cognitive impairment and early Alzheimer’s disease. J Comp Neurol. 1999; 411: 693- 704.
- De Kosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002; 51: 145-155.
- Ikonomovic MD, Mufson EJ, Wuu J, Bennett DA, DeKosky ST. Reduction of choline acetyltransferase activity in primary visual cortex in mild to moderate Alzheimer’s disease. Arch Neurol. 2005; 62: 425-430.
- Hara Y, Motoi Y, Hikishima K, Mizum H, Onoe H, Matsumoto SE, et al. Involvement of the septo-hippocampal cholinergic pathway in association with septal acetylcholinesterase upregulation in a mouse model of tauopathy. Curr Alzheimer Res. 2016.
- Konishi K, Hori K, Tomioka H, Minegishi G, Tani M, Tanaka H, et al. Donepezil abolishes anticholinergic activity in a patient with amnesia. Pharmacology. 2013; 91: 86-91.
- Tune L, Carr S, Hoag E, Cooper T. Anticholinergic effects of drugs commonly prescribed for the elderly: potential means for assessing risk of delirium. Am J Psychiatry. 1992; 149: 1393-1394.
- Flacker JM, Lipsitz LA. Serum anticholinergic activity changes with acute illness in elderly medical patients. J Gerontol A Biol Sci Med Sci. 1999; 54: M12-M16.
- Plaschke K, Kopitz J, Mattern J, Martin E, Teschendorf P. Increased cortisol levels and anticholinergic activity in cognitively unimpaired patients. J Neuropsychiatry Clin Neurosci. 2010; 22: 433-441.
- Hori K, Hachisu M, Tomioka H, Konishi K. Anticholinergic Activity and Alzheimer’s Disease. Neurodegener Dis. 2015; 15: 131-133.
- Darvesh S, Hopkins DA, Geula C. Neurobiology of butyrylcholinesterase. Nat Rev Neurosci. 2003; 4: 131-138.
- Horiuchi K, Hori K, Hosoi M, Konishi K, Tomioka H, Hachisu M. Rivastigimine for Relatively Younger Alzheimer’s Disease Patient. Brain Disord Ther. 2014, 3:133.
- Hori K, Konishi K, Tomioka H, Tani M, Minegishi G, Tanaka H, et al. Galantamine for aggressive behavior in Alzheimer’s disease. J New Remedies Clinics 2012; 61: 1304-1305.