
Case Report
Austin J Clin Neurol 2021; 8(1): 1146.
Adult-onset Neurological Langerhans Cell histiocytosis and Response to Treatment
Al-Hader R1, Suneja A1, Mukherje A2 and Memon AB1,3*
¹Department of Neurology, Henry Ford Hospital, USA
²Department of Pathology, Henry Ford Hospital, USA
³Wayne State University, School of Medicine, USA
*Corresponding author: Anza Bilal Memon, Department of Neurology, Henry Ford Hospital, 2799 W. Grand Blvd., Detroit, Michigan 48202, USA
Received: April 12, 2021; Accepted: April 28, 2021; Published: May 05, 2021
Abstract
Introduction: Langerhans Cell Histiocytosis (LCH) is a rare form of cancer that mostly affects children and rarely adults. LCH involves an abnormal clonal proliferation of Langerhans cells in the bone marrow. These cells are capable of migrating from the skin to lymph nodes. Therefore, it is characterized as a multisystem disease. Neurological manifestations are not common, and often patients’ present with endocrine dysfunction with neuroimaging findings of hypothalamic and pituitary masses can mimic pituitary adenoma. Here, we discuss two instances of unusual adult-onset, primary neurological LCH in patients with a positive response to therapy-these two patients presented with mass lesion and neurodegenerative form of LCH, respectively. LCH can manifest features of mass lesions or neurodegeneration on brain Magnetic Resonance Imaging (MRI). Since it is rare in adults, it is crucial to identify this condition as timely treatment can have a better prognosis.
Keywords: LCH; Pituitary tumor; Neurodegeneration; Central diabetes insipidus
Introduction
Langerhans Cell Histiocytosis (LCH) is a neoplasm of myeloid origin characterized by clonal proliferation and dissemination of cells that express the CD1a and CD207 cell surface proteins. Up to 75% of LCH patients have sequence variations in the BRAF and MAP2K1 genes, contributing to the constitutive activation of the ERK signaling pathways which is a feature of LCH [1]. LCH typically is a multiorgan disease that affects children younger than 15 years; however, adult-onset LCH can appear after the fourth decade of life, often leading to delayed diagnosis.
LCH with CNS involvement can manifest in a variety of forms [2]. It can present with Diabetes Insipidus (DI), neurodegenerative changes, and mass lesions [3-7]. The most common manifestation of LCH is the DI, followed by the neurodegenerative changes [3-7]. Mass lesions are the least common CNS manifestation of LCH [3,4]. These lesions can arise from meninges (dura matter), hypothalamus, pineal gland and choroid plexus. The neurodegenerative changes are typically seen in the posterior fossa and can involve the cerebellum, brainstem and basal ganglia [3-7].
Hypothalamic pituitary involvement, and DI is seen in about 25% of the patients with LCH and as many as 50% of patient with multisystem disease [8]. LCH is generally characterized by a wide range of symptoms that vary depending on the organ involved, with the most common symptoms being bone pain and skin rash. Nevertheless, idiopathic central diabetes insipidus, large pituitary tumors, and anterior pituitary dysfunction in otherwise healthy adults indicate LCH with CNS involvement. In this case series, we describe two patients who presented with adult-onset CNS manifestations of LCH with a mass lesion that involved the pituitary-hypothalamic axis and neurodegenerative form the disease. Both patients had a good response to treatment.
Case Presentati
Case 1: A 43-year-old woman presented with recurrent syncopal episodes, daily headaches, difficulty with word choice, visual field deficits, and short-term memory problems. Upon examination, she was noted to have bitemporal hemianopsia. Magnetic Resonance Imaging (MRI) of the brain revealed a 23.4 x 21.6 x 15.3 mm suprasellar mass with mild inferior displacement of the optic chiasm associated with vasogenic edema. Fluid-Attenuated Inversion Recovery (FLAIR) images showed a potential mass effect associated with gadolinium enhancement without diffusion restriction (Figure 1, A-C). The patient underwent bifrontal craniotomy and suprasellar mass debulking. Histological test results revealed a proliferation of histiocytes admixed with lymphocytes, plasma cells, and some eosinophils. Histiocytes had a moderate amount of foamy cytoplasm, which is indicative of LCH (Figure 1, G-I). Post-operatively, the patient presented with panhypopituitarism and central diabetes insipidus. Positron Emission Tomography (PET) showed hypermetabolic left cervical lymph nodes; however, the lymph nodes’ fine-needle aspiration did not reveal typical LCH pathology. The patient started radiation therapy. The patient remained clinically stable, and an MRI of the brain one year later showed a suprasellar mass that was slightly smaller relative to initial imaging (Figure 1, D-F). The patient is being followed in the outpatient neurology, endocrinology, ophthalmology, and oncology clinic for close observation.
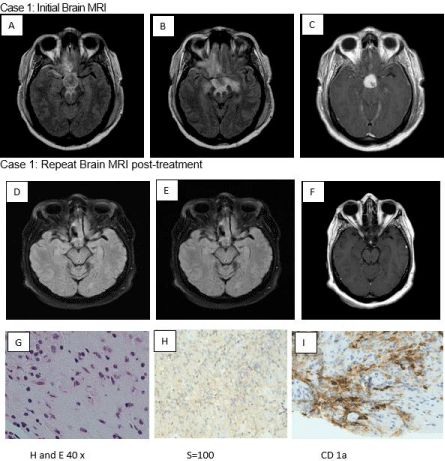
Figure 1: FLAIR axial (A,B) shows T2 Hyperintensity in the bilateral orbitofrontal gyrus, pituitary stalk, neural hypophysis, and optic chiasm associated with
Gadolinium (Gd) enhancement (C). Repeat Brain MRI FLAIR axial shows minimally smaller suprasellar mass (D,E,F).
Pathology pictures show proliferation of histiocytes admixed with lymphocytes, plasma Cells and occasional eosinophils. The histiocytes have a moderate amount
of foamy cytoplasm ((H and E stain). The histiocytic cells are focally immunoreactive to S-100 and CD1a (G,H,I).
Case 2: A 28-year-old man presented with significant gait unsteadiness, diplopia, nystagmus, dysphagia, and dysarthria. MRI of the brain with and without contrast revealed FLAIR signal abnormalities in the pons (brainstem) and cerebellum without associated enhancement or diffusion restriction (Figure 2, A-D). The patient was diagnosed with neurodegenerative LCH with panhypopituitarism based on neuroimaging findings. A biopsy could not be performed because of the sensitive location of the brain lesions. He underwent 12 cycles of cytarabine treatment (170mg/ m²) at the time of presentation and 6 cycles of cytarabine 6 months later. After treatment, his symptoms decreased, and his condition improved. The patient is currently clinically stable but has residual neurological deficits, including horizontal gaze-evoked nystagmus on lateral gaze (worse to the right) and mild gait ataxia A repeated MRI of the brain 11 years after initial presentation showed resolution of T2 signal changes in the pontine and cerebellar regions. However, interval advanced pontine and cerebellar atrophy was also identified (Figure 2 E).
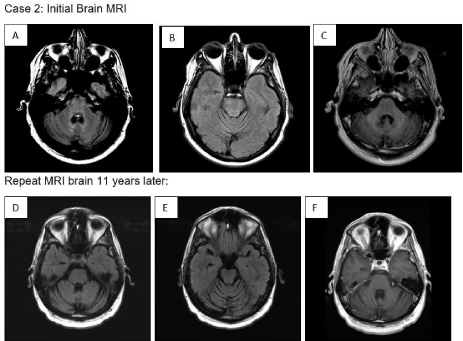
Figure 2: 2: FLAIR axial (A) shows T2 hyperintensity present within the pons, superior cerebellar peduncles, and within the cerebellum adjacent to the fourth ventricle
in the region of the dentate nuclei with mild cerebellar atrophy (B), without associated enhancement (C).
Repeat MRI brain 11 years later shows interval resolution of the T2 hyperintensity on FLAIR sequences within the pons and cerebellum (D,E) without GD
enhancement (F). However, it shows significant interval pontine and cerebellar atrophy (E).
Discussion
Only 6% of patients with LCH can present with neurological manifestations at the onset of diagnosis [9]. There are three distinct CNS manifestations of LCH that may or may not be present simultaneously in the same patient, such as central or neurogenic DI, obstructive hydrocephalus, and neurodegenerative LCH characterized by gait, extremity ataxia, and psychomotor deterioration [3-5].
The approach to treating LCH can be tailored by explicitly considering the involved organ. Treatments may range from surgical curettage to systemic chemotherapy, but evidence and guidelines regarding customized treatments for neurodegenerative LCH of the CNS are limited. The current treatment options for LCH are transretinoic acid, intravenous immunoglobulin, steroids (with or without vinblastine, mercaptopurine, or methotrexate), and cytarabine (with or without vincristine) [10,11]. Upon imaging, LCH is usually associated with T2 hyperintense gadolinium-enhancing lesions, most commonly involving the pineal gland, pituitary stalk, and dentate nucleus [12]. PET imaging should be performed for all patients with suspected histiocytic neoplasms to assess organ involvement.
The two unusual cases of adult-onset, primary neurological LCH in patients who had a positive response to therapy described here should help physicians identify and treat this rare condition. Physicians should consider LCH within the differential diagnosis for patients who have progressive pituitary dysfunction. Examining clinical, radiologic, and histopathologic findings together is critical for establishing a neuroendocrine diagnosis of LCH. Early recognition and prompt, appropriate treatment strategies are crucial to prevent the progression of LCH and avoid progression to early disability.
Acknowledgment
The authors thank Karla D Passalacqua, PhD, at Henry Ford Hospital for editorial assistance.
References
- Hutter C, Minkov M. Insights into the pathogenesis of langerhans cell histiocytosis: The development of targeted therapies. ImmunoTargets Ther. 2016.
- Monsereenusorn C, Rodriguez-Galindo C. Clinical Characteristics and Treatment of Langerhans Cell Histiocytosis. Hematol Oncol Clin North Am. 2015.
- D’Ambrosio N, Soohoo S, Warshall C, Johnson A, Karimi S. Craniofacial and intracranial manifestations of langerhans cell histiocytosis: report of findings in 100 patients. AJR. American journal of roentgenology. 2008; 191: 589-597.
- Prayer D, Grois N, Prosch H, Gadner H, Barkovich AJ. MR Imaging Presentation of Intracranial Disease Associated with Langerhans Cell Histiocytosis. American Journal of Neuroradiology. 2004; 25: 880-891.
- Zaveri J, La Q, Yarmish G, Neuman J. More than just Langerhans cell histiocytosis: a radiologic review of histiocytic disorders. Radiographics: a review publication of the Radiological Society of North America, Inc. 2014; 34: 2008-2024.
- Chaudhary V, Bano S, Aggarwal R, Narula MK, Anand R, Solanki RS, et al. Neuroimaging of Langerhans cell histiocytosis: a radiological review. Japanese journal of radiology. 2013; 31: 786-96.
- Laurencikas E, Gavhed D, Stålemark H, van’t Hooft I, Prayer D, Grois N, et al. Incidence and pattern of radiological central nervous system Langerhans cell histiocytosis in children: a population based study. Pediatric blood & cancer. 2011; 56: 250-257.
- Bernstrand C, Sandstedt B, Ahström L, Henter JI. Long-term follow-up of Langerhans cell histiocytosis: 39 years’ experience at a single centre. Acta Paediatr. 2005; 94: 1073-1084.
- Grois N, Pötschger U, Prosch H, Minkov M, Arico M, Braier J, et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatric blood & cancer. 2006; 46: 228-233.
- Allen CE, Flores R, Rauch R, Dauser R, Murray JC, Puccetti D, et al. Neurodegenerative central nervous system Langerhans cell histiocytosis and coincident hydrocephalus treated with vincristine/cytosine arabinoside. Pediatr Blood Cancer. 2010.
- Imashuku S, Okazaki N, Nakayama M. Treatment of neurodegenerative CNS disease in langerhans cell histiocytosis with a combination of intravenous immunoglobulin and chemotherapy. Pediatr Blood Cancer. 2008.
- Porto L, Schöning S, Hattingen E, Sörensen J, Jurcoane A, Lehrnbecher T. Central nervous system imaging in childhood Langerhans cell histiocytosis - A reference center analysis. Radiol Oncol. 2015.