
Research Article
Austin J Clin Ophthalmol. 2014;1(5): 1023.
Increased ER Stress as a Mechanism of Retinal Neurovasculopathy in Mice with Severe Hyperhomocysteinemia
Amany Tawfik1,2 and Sylvia B Smith1,2,3*
1Department of Cellular Biology and Anatomy, Georgia Regents University, USA
2James and Jean Culver Vision Discovery Institute, Georgia Regents University, USA
3Department of Ophthalmology, Medical College of Georgia, Georgia Regents University, Augusta, USA
*Corresponding author: Sylvia B Smith, Department of Cellular Biology and Anatomy, Medical College of Georgia, Georgia Regents University, 1120 15th Street, CB 1114, Augusta, 30912-2000, GA, USA
Received: May 21, 2014; Accepted: June 13, 2014; Published: June 16, 2014
Abstract
Hyperhomocysteinemia is implicated in retinal neurovascular diseases including arterial occlusive disease, venous occlusive disease and pseudoexfoliation glaucoma. The mechanism for these diseases is not known. Here we used hyperhomocysteinemic mice lacking the gene encoding cystathionine-beta-synthase (cbs -/-) to examine whether ER stress could be a mechanism for the retinal neurovasculopathy reported in these mice. Retinas of cbs+/+and cbs-/- mice (age: 3-5 wks) were used to investigate the expression of ER stress genes (BiP/GRP78, Perk, Atf6, Atf4, Ire1α,Chop) and the proteins they encode. The levels of poly (ADP-ribose) Polymerase (PARP) and cleaved cysteine-aspartic proteases-3 (caspase-3), proteins known to be involved in apoptosis, were also examined. Quantitative reverse transcription polymerase chain reaction and western blotting revealed an increase in BiP/ GRP78 and PERK in retinas of cbs -/-mice compared with cbs+/+mice. There was an elevation of CCAAT-enhancer-binding protein Homologous Protein (CHOP) in retinal cryosections of cbs -/- mice indicating apoptosis, which was confirmed by increased levels of PARP and cleaved caspase-3. The data suggest that the genes and proteins that are major players in the ER stress pathway, particularly the PERK pathway, are up regulated in retinas of cbs -/- mice. The data support a role for ER stress in the pathophysiology associated with the hyperhomocysteinemia-linked retinal disease.
Keywords : Homocysteine; Retinal degeneration; Retinal neurovasculopathy; Apoptosis; BiP/GRP78; Cystathionine-β-synthase
Abbreviations
ER: Endoplasmic Reticulum; Hcy: Homocysteine; HHcy: Hyperhomocysteinemia; SAM: S-Adenosylmethionine; CBS: Cystathionine Beta Synthase; MTHFR: Methylene Tetrahydrofolate Reductase; NMDA: N-methyl-D-aspartate Receptor; VEGF: Vascular Endothelial Growth Factor, UPR: Unfolded Protein Response; BiP/ GRP78: Immunoglobulin Heavy Chain-binding Protein; CHOP: CCAAT-enhancer-binding Protein Homologous Protein; PERK: PKR-like Endoplasmic Reticulum Kinase; IRE1: Inositol-requiring Enzyme 1; ATF6: Activating Transcription Factor 6; ATF4: Activating Transcription Factor 4; qRT-PCR: Quantitative Reverse Transcription Polymerase Chain Reaction; HRP: Horseradish Peroxidase
Introduction
This study investigated Endoplasmic Reticulum (ER) stress as a mechanism of homocysteine-linked retinopathy. Homocysteine (Hcy), a key intermediate in metabolism of methionine, is the direct precursor of S-Adenosyl Methionine (SAM), the most important methyl group donor in the body. Depending upon metabolic demands, Hcy is the substrate for regeneration of methionine (remethylation pathway) or is diverted from the methionine cycle to produce cystathionine and ultimately cysteine (transsulfuration pathway). Normal human fasting plasma levels of Hcy (total Hcy) are 5-15μmol/l; while Hyperhomocysteinemia (HHcy) is classified as: moderate (16-30μmol/l), intermediate (31-100μmol/l), severe (>100μmol/l) [1]. HHcy is an independent risk factor in cardiovascular diseases (stroke, venous thrombosis, peripheral arterial occlusive disease [1-4]) and neurodegenerative diseases (Alzheimer’s and Parkinson’s Diseases) [5,6]. Given that the retina is a neurovascular tissue, it is not surprising that many clinical studies have investigated levels of Hcy in retinopathies [7,8]. HHcy is a risk factor in retinal venous and arterial occlusions [9] including central retinal vein occlusion [10-14]. It has been linked to pseudoexfoliation glaucoma [15-19], macular degeneration [20-22] and diabetic retinopathy [23- 25].
HHcy is caused by genetic deficiencies in enzymes responsible for transsulfuration or remethylation of Hcy or by nutritional deficiencies in vitamins serving as cofactors for these enzymes (folate/B12 /B6). The most frequent genetic deficiencies are associated with the enzymes Cystathionine Beta Synthase (CBS) and Methylene Tetrahydrofolate Reductase (MTHFR). There are more than 150 disease-associated CBS mutations, making this the most common genetic cause of severely elevated Hcy [26,27]. The I278T mutation accounts for 25% of all homocystinuric alleles, and is a common cause of Homocystinuria (severe HHcy), a genetically inherited, autosomal recessive inborn error of metabolism (1:200,000 live U.S. births) [28]. The cornerstone clinical features of these patients are mental retardation, ectopia lentis, visual deficits and skeletal abnormalities; premature death istypically due to thromboembolic events [29].
Our lab and others have performed in vitro and in vivo studies to elucidate mechanisms of HHcy-linked retinal disease. A clinically relevant experimental system is the mouse deficient or lacking the gene encoding CBS allowing studies of the effects of mild to severe endogenous elevation of Hcy [30]. In previous studies, we examined consequences on retina structure and function using either cbs-/-mice, which have a 30-40 fold increase in plasma Hcy and a shortened life-span of 3-5 weeks; or cbs+/-mice, which have a much milder HHcy with ~4-7 fold increase in plasma Hcy (and a 2-fold increase in retinal Hcy) and a normal lifespan. Our work has shown that both cbs-/-and cbs+/-mice have retinal neuronal involvement and disruption of the retinal vasculature [31-36].
To understand mechanisms for HHcy-induced retinal neuronal death we previously investigated the role of excitotoxicity and oxidative stress using perforated patch clamp analysis and fluorescent detection of intracellular Ca2+ in primary mouse retinal ganglion cells and found that Hcy-induced cell death, which was blocked partially by MK-801, an N-methyl-D-aspartate receptor (NMDA) receptor antagonist [36]. Hcy increased intracellular Ca2+ 7-fold. Additionally exposure of ganglion cells to 50 μM Hcy increased levels of superoxide, nitric oxide and peroxynitrite levels by 40%, 90% and 85%, respectively. We also investigated retinal vasculature in mice with HHcy and observed a marked vasculopathy developing very early in cbs-/- mice associated with increased levels of Vascular Endothelial Growth Factor (VEGF) [33]. Interestingly, VEGF has been linked to ER stress in various systems including retina [37-39], prompting this investigation of well-known ER stress markers. We were interested in determining whether ER stress plays a role in Hcy-induced retinal neurovascular pathology observed incbs-/- mice.
Materials and Methods
Animals
The generation of mice deficient in cbs has been reported [30]. Breeding pairs of cbs+/- mice (B6.129P2-Cbstm1Unc/J; Jackson Laboratories, Bar Harbor, ME) were used to establish our mouse colony. Genotyping, husbandry and housing conditions for the mice have been described [33]. Wild type (cbs+/+, n = 17) and homozygous mutant (cbs-/-, n = 18) mice were used in this study at ~3 weeks. Mean body weight for cbs+/+ mice (6.84 ± 0.2 g) was significantly greater than age-matched cbs-/- mice (4.53 ± 0.2 g).Experiments adhered to the Association for Research in Vision and Ophthalmology Statement forthe Use of Animals in Ophthalmic and Vision Research and followed our animal use protocol approved by the Institutional Animal Care and Use Committee of Georgia Regents University.
Analysis of VEGF levels in retinas of severely hyperhomocysteinemic mice
Elevation of VEGF has been linked to increased ER stress in various tissues, this observation, along with other evidence suggesting ER stress as a mechanism of HHcy-induced pathology, prompted the current experiments. We performed immunohistochemical studies in cryosections using mouse monoclonal anti-VEGF (IgM) (1:250, Abcam Corp., Cambridge, MA) and detected the protein using Alexafluor 488 (donkey anti-mouse antibody, Invitrogen, Eugene, OR).The Metamorph analysis system was used to quantify immunofluorescence levels.
Real time quantitative RT-PCR (RT-q PCR) analysis of genes in the ER stress pathways
Expression levels of mRNA transcripts specific for several key genes involved in ER stress pathways (BiP/GRP78, Perk, Atf6, Ire1α, Atf4, Chop) were examined in neural retinas isolated from cbs+/+ and cbs-/- mice per our method [40]. Total RNA was isolated using TRIzolTM Reagent (Invitrogen, Carlsbad, CA) and quantified. 2 μg of RNA was reverse transcribed using iScript™ Synthesis kit (BioRad Laboratories, Hercules, CA). cDNAs were amplified for 45 cycles using Absolute QPCR SYBR Green Fluorescein (AB gene, Surrey, UK) and gene specific primers (Table 1) using the Bio-Rad icycler (Hercules, CA). Expression levels were calculated by comparison of Ct values (delta-delta Ct).PCR was performed (40 cycles: 95°C for 30 s; 60°C for 30 s; 72°C for 30 s); melt curve analysis confirmed end product purity. Resulting CT values were normalized to 18s and analyzed using the comparative CT method to obtain fold-changes in gene expression.
![]()
Gene
NCBI Accession No.
Primer Sequence
Predicted band size
BiP
NM_022310
Forward: 5’-ACTTGGGGACCACCTATTCCT-3’
Reverse:� 5’-ATCGCCAATCAGACGCTCC-3’
134
Perk
NM_010121
Forward: 5’-AGTCCCTGCTCGAATCTTCCT-3’
Reverse:� 5’-TCCCAAGGCAGAACAGATATACC-3’
125
Atf4
NM_009716
Forward: 5’-TCCTGAACAGCGAAGTGTTG-3’
Reverse:� 5’-ACCCATGAGGTTTCAAGTGC-3’
129
Ire1a
NM_023913
Forward: 5’-ACACCGACCACCGTATCTCA-3’
Reverse:� 5’-CTCAGGATAATGGTAGCCATGTC-3’
110
Atf6
NM_001107196
Forward: 5’-TGCCTTGGGAGTCAGACCTAT-3’
Reverse: 5’-GCTGAGTTGAAGAACACGAGTC-3’
141
Chop
NM_007837
Forward: 5’-CTGGAAGCCTGGTATGAGGAT-3’
Reverse: 5’-CAGGGTCAAGAGTAGTGAAGGT-3’
121
18S
NR_003278
Forward: 5’-AGTGCGGGTCATAAGCTTGC-3’
Reverse: 5’-GGGCCTCACTAAACCATCCA-3’
90
Table 1: Overview of autoimmune disease in whose development Mast cells may be involved.
Immunoblotting to detect levels of major ER stress proteins
Proteins were extracted from neural retinas isolated from cbs+/+and cbs-/-mice to detect BiP/GRP78 (immunoglobulin heavy chain-binding protein, a 78 kD glucose regulated protein) (BD Bioscience, San Jose, CA), ATF6 (Activating Transcription Factor-6),CHOP(C/EBP homologous protein) (Santa Cruz Corp., Santa Cruz, CA), PERK(RNA-dependent Protein Kinase (PKR)-like ER kinase), p-PERK (Phosphorylated PERK),cleaved caspase-3 (cysteine-aspartic proteases-3), PARP (poly(ADP-ribose) polymerase)(Cell Signaling, Danvers, MA)per our published method [33]. Protein samples were subjected to SDS-PAGE and transferred to nitrocellulose membranes, which were incubated with the above antibodies at 4oC overnight, followed by a Horseradish Peroxidase (HRP)-conjugated goat anti-mouse IgG (1:5000) or goat anti-rabbit IgG antibody (1:3000). Nitrocellulose membranes, to which the proteins had been transferred, were incubated with primary antibodies at a concentration ranging from 1:100 to 1:500. They were incubated with HRP-conjugated goat anti-rabbit (Santa Cruz Corp., 1: 3000) or goat anti-mouse IgG antibody (Sigma-Aldrich, St. Louis, MO, 1:3000). Proteins were visualized using the Super Signal West Pico Chemiluminescent Substrate detection system (Pierce Biotechnology, Rockford, IL). Membranes were reprobed with mouse monoclonal anti-β-actin antibody (1:5000) as a loading control. Protein levels were quantified as described [33].
Immunofluorescent assessment of ER stress proteins in retinal cryosections
The ER stress protein BiP/GRP78 as well as CHOP and cleaved caspase-3 were detected using immunofluorescence in cryosections prepared from cbs+/+ and cbs-/- mice per our method [33]. Cryosections were fixed with 4% paraformaldehyde, incubated with Power Block followed by incubation with primary antibody for either 3 h at 37oC or overnight at 4°C. Sections were incubated with secondary antibody and covers lipped with Fluoro shield with DAPI (Sigma-Aldrich) to label nuclei. They were visualized by immunofluorescence using an Axioplan-2 fluorescent microscope (Carl Zeiss, Göttingen, Germany) equipped with a High Resolution Microscope (HRM) camera. Images were captured and processed using Zeiss Axiovision digital image processing software (version 4.7). The Metamorph analysis system was used to quantify immunofluorescence levels. Following our published method [41], neurons of the ganglion cell layer were labeled with NeuN (Neuronal Nuclei, red fluorescence) and Brn-3a (green fluorescence) and were counted to determine the number of neurons in this layer in retinas of cbs+/+ and cbs-/- mice. Images werecaptured from the central, mid-peripheral and peripheral retina on both sides of the optic nerve (6 images from each retinal section); we examined 3 sections per slide and 3 slides per retina. Three mice per group were used in this analysis.
Statistical analysis
qPCR and densitometric scans for western blotting of BiP/GRP78, ATF6, ATF4, IRE1, PERK, pPERK, CHOP, cleaved caspase-3, cleaved PARP as well as metamorphic quantification of immunofluorescent detection of BiP/GRP78, CHOP, VEGF, cleaved caspase-3 were analyzed using the Graph Pad Prism software (La Jolla, CA). Tests used included unpaired t-test, F test for unequal variance, paired t-test and Student’s test. A p value <0.05 was considered significant.
Results
Confirmation that VEGF levels increase in retinas of cbs -/- mice
Increased VEGF levels are linked to increased ER stress. In the current study, we confirmed our previous findings that VEGF levels are increased in retinas of cbs-/-mice [33]. Immunofluorescent detection of VEGF in retinal cryosections revealed marked increase in VEGF in retinas of cbs+/+ mice compared with cbs+/+ mice (Figure 1A). In studies in which the primary antibody was omitted (negative control) or when IgM was used in place of the primary antibody (isotype control) there was no signal detected (data not shown). Metamorph analysis of the color intensity of VEGF labeled cryosections revealed a marked increase in levels in the cbs-/- retinas compared to wild type retinas Figure 1B. The data reaffirm our earlier findings [33] that VEGF levels increase in retinas under conditions of severe hyperhomocysteinemia and set the stage for analysis of ER stress in this model.
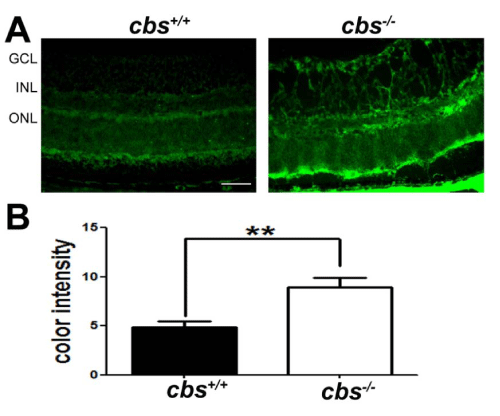
Figure 1 :Detection of VEGF in in retinas of cbs+/+and cbs-/- mice. Retinal cryosections from cbs+/+and cbs-/-mice were subjected to immunofluorescent detection of VEGF (green fluorescence), a marker of angiogenesis. (B) Quantification of the data obtained from metamorphic analysis of color intensity of VEGF (**p<0.01, n=6). (Calibration bar: 50μm) Abbreviations: GCL = ganglion cell layer, INL = inner nuclear layer, ONL = outer nuclear layer.
Analysis of ER stress genes and proteins in retinas of cbs-/- mice
There have been a number of studies implicating ER stress as a mechanism mediating Hcy-induced atherosclerosis [reviewed in 42]. In retina, there have been many studies implicating ER stress in retinal degenerations [reviewed in 43], however few linking Hcy and ER stress in retinal disease. There was a report that excess Hcy increased VEGF via ER stress in vitro and increased transcription of ATF4 [44], but no studies that examine ER stress in retinas with endogenously elevated Hcy. We explored the in vivo role of Hcy in modulating retinal expression of major ER stress genes including BiP/GRP78and its downstream effector genes (Perk, Atf6, Ire1α), by analyzing their expression in neural retina of cbs-/- mice compared to wild type mice. qRT-PCR revealed a significant increase in the expression of BiP/ GRP78 and Perk (Figure 2) in the cbs-/- mouse retina compared with wild type mice. Interestingly, we observed a trend toward increased levels of ATF6 and ATF4, although the data did not reach statistical difference.
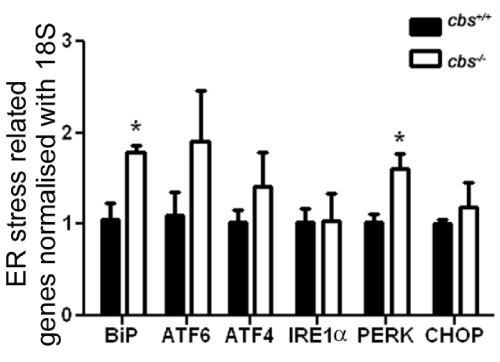
Figure 2:Analysis of genes encoding the ER stress effector proteins in the neural retina of cbs+/+ and cbs-/- mice. mRNA of the neural retina was isolated from cbs+/+ and cbs-/- at age ~3-5 weeks old. Real time qRT-PCR was performed to analyze the expression of BiP/GRP78, ATF6, ATF4, IRE1α, PERK, p-PERK and CHOP/GADD34. Levels were normalized to 18S. Each experiment was performed in triplicate;*p <0.05.
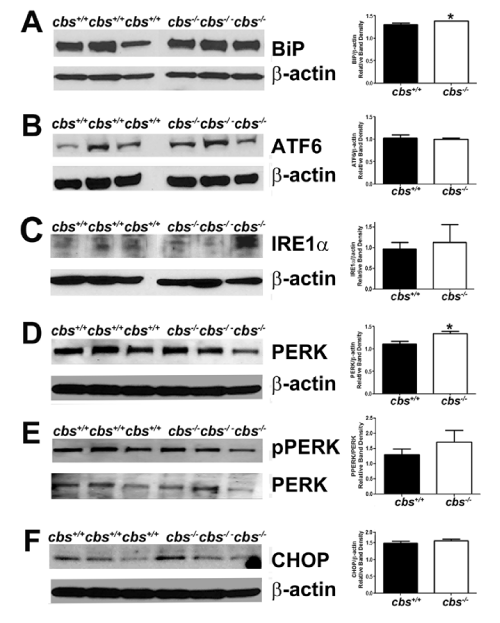
Figure 3 : Analysis of major ER stress proteins in the neural retina of cbs+/+and cbs-/-mice. Proteins from neural retinas of cbs+/+and cbs-/-mice were isolated and subjected to immunoblotting to detect major proteins implicated in the ER stress response: (A) BiP, (B) ATF6, (C) IRE1α, (D) PERK, (E)p-PERK, (F) CHOP. Band densities were normalized to β-actin and densitometric analysis of the bands are provided next to each blot. (*p<0.05).
We examined the expression of the proteins encoded by major ER stress genes Fig. 3A-F). The protein level was expressed as a ratio of band densities of the protein of interest compared to the level of the loading control (β-actin). The analysis of proteins of cbs-/- mice revealed a significant increase in the level of BiP/GRP 78 compared with wild type (mean ± SEM: 1.386 ± 0.002 (cbs-/-)v. 1.308±0.029 (cbs+/+)) (Fig. 3A). There was a significant increase also in the levelof PERK in the cbs-/- mice compared with cbs+/+ mice (mean ± SEM: 1.336 ± 0.0535 (cbs-/-) v. 1.107 ± 0.061(cbs+/+)) (Fig. 3D). Taken collectively, there appears to be an increase in the major regulator of ER stress as well as involvement of the PERK pathway for ER stress in retinas of HHcy mice.
Cellular localization of BiP/GRP78 and CHOP: evidence of apoptosis in retinas of cbs-/- mice
We next asked in which retinal cell layers were the increased BiP/ GRP78 protein present in cbs-/- mice. There was a marked increase in the level of BiP/GRP78 in the retinas of cbs-/- compared with cbs+/+ mice (Figure 4A), which was quantified and found to be significant (Figure 4B). The inner retinal layers, especially the ganglion cell layer, appeared to have greater levels of BiP/GRP78 than did the outer retinal cells. It is well known that if ER stress cannot be suppressed through the cellular machinery for correcting misfolded proteins, then apoptosis will be triggered so that the cell will die allowing the remaining tissue to survive [43,45]. In the ER, apoptosis is signaled through several mechanisms including a PERK-dependent pathway that can cause induction of CHOP. Given that PERK levels were increased in retinas of cbs-/- compared with cbs+/+ mice (Figure 2 and 3), we examined CHOP in the retinas of these mice. We used immunohistochemical methods to detect CHOP in retinal cryosections (Figure 4C, D) and found that CHOP levels were markedly increased in the retinas of cbs-/- compared with cbs+/+ mice. As with BiP/GRP78, the inner retina, particularly ganglion cells, and the radially oriented Muller cells appeared to have the most intense expression of CHOP. These data prompted analysis of the numbers of cells in the ganglion cell layer of cbs-/- compared with cbs+/+ mice. To investigate this, we labeled retinal cryosections with the neuronal marker NeuN and the ganglion cell marker Brn-3 using immunohistochemical methods (Figure 4E). We then counted the number of cells in the ganglion cell layer in the two mouse groups following our published method [41]. The data showed a significant decrease in the number of cells in the ganglion cell layer of the cbs-/- mice compared with age-match wild type mice (Figure 4F).
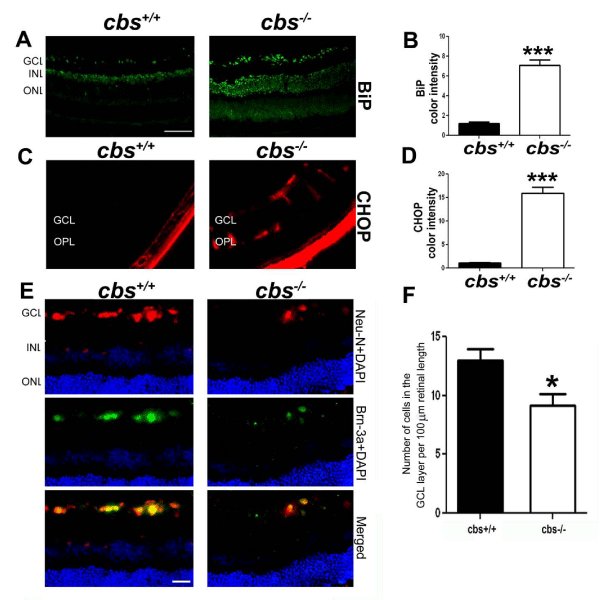
Figure 4 : Immunohistochemical detection of BiP and CHOP in retinas of cbs+/+and cbs-/-mice and evidence of decreased numbers of retinal ganglion cells in cbs-/- mice. Retinal cryosections from cbs+/+ and cbs-/-mice were subjected to immunofluorescent detection of (A) BiP (green fluorescence) and (C) CHOP (red fluorescence). The color intensity of BiP (B) and CHOP (D) were quantified using the Metamorph analysis system. (Calibration bar: 50μm, ***p<0.001, n = 6). (E) Neurons of the ganglion cell layer were labeled with neuN (red fluorescence) and Brn-3a (green fluorescence) and were counted to determine the number of neurons in retinas of cbs+/+ and cbs-/- mice; data were depicted graphically (F). (***p<0.001, * p<0.05, n = 6; calibration bar: 50μm,).
CHOP was initially reported as a transcription factor involved in ER stress-induced apoptosis [15]. We explored apoptosis in retinas of cbs-/- mice by examining levels of PARP and cleaved caspase-3. PARP catalyzes the poly(ADP-ribosyl) ation of a variety of nuclear proteins with Nicotinamide Adenine Dinucleotide (NAD) as substrate. It is activated by binding to DNA ends, however when it is cleaved it becomes inactive and can no longer respond to DNA strand breaks [46,47]. The specific cysteine protease that plays a central role in execution of the apoptotic program is caspase-3 [46]. We found a significant increase in the level of cleaved caspase-3 in retinas of the cbs-/- mice (Figure 5A, B) and a marked increase in the level of cleaved PARP (Figure 5C,D). In retinal cryosections, there were significantly more cleaved caspase-3 positive cells in retinal cryosections (Figure 5E,F) of cbs-/-mice compared to cbs+/+ mice. Interestingly, some of the cleaved caspase-3 cells co-localized with isolectin-B4, a marker for blood vessels.
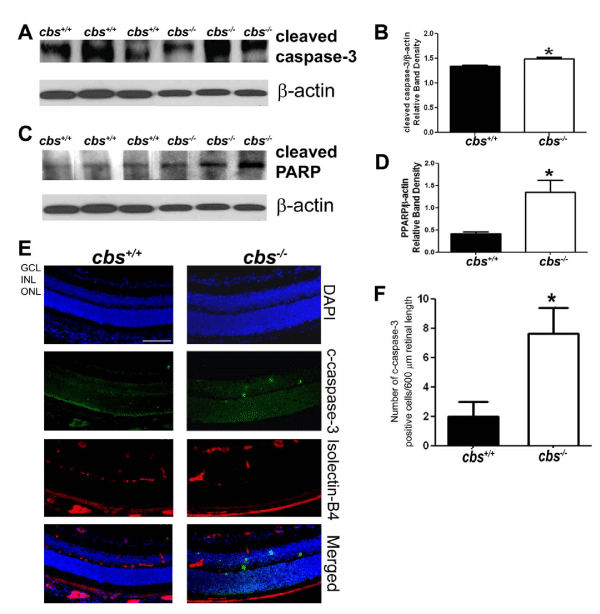
Figure 5 : Evaluation of cleaved caspase-3 and PARP in retinas of cbs+/+and cbs-/- mice. Proteins from neural retinas of cbs+/+and cbs-/-mice were isolated and subjected to immunoblotting to evaluate the levels of apoptotic markers cleaved caspase-3 (A, B) and cleaved PARP (C,D). Band densities were normalized to β-actin and densitometric analysis of the bands are provided next to each blot. (E) Panel of retinal cryosections from cbs+/+and cbs-/-mice subjected to immunofluorescence methods to detect cleaved caspase-3 and retinal blood vessels in retinal cryosections (blue = DAPI to label nuclei, green = detection of cleaved caspase-3, red = detection of retinal vessels using isolectin-B4). (F) The number of cleaved caspase -3 positive cells were counted in cryosections (*p <0.05; calibration bar: 50μm).
Discussion
The present study examined ER stress in the retinal neurovasculopathy observed in mice with severely elevated levels of Hcy. HHcy is relevant to human retinal neurovasculopathies, including retinal vein occlusion, retinal artery occlusion and pseudoexfoliation glaucoma [7-19]. Clinical studies are often inconclusive with respect to mechanisms underlying disease. In vitro studies, in which neuronal or vascular cell types are incubated with varying concentrations and formulations of Hcy, provide some clues about pathological mechanisms, although endogenously occurring models are likely to provide insights that will be more relevant to human pathophysiology. For these reasons, we have been investigating mechanisms by which moderate to severe endogenous elevation of Hcy may alter the neurons or vessels of the retina and have used mouse models that have genetic defects in the Hcy metabolic pathway.
The cbs-/-mouse used in this study is an example of very severely elevated plasma Hcy (and ~7-fold elevation of retinal Hcy). The marked neuronal death and vascular pathology observed in the retina are evident within the first three weeks of life [31,33,35]. The model has provided an excellent opportunity to investigate mechanisms ofHcy-induced retinal neurovasculopathy over a very short time frame, in this case 3-5 weeks. The cbs+/-mouse, which is less severe HHcy, has also proven useful in mechanistic studies of Hcy-induced retinal disease [31,32,34,36,48].
ER stress is a fundamental cellular process. Typically, proteins are translocated into the ER lumen in an unfolded state and require protein chaperones/catalysts of protein folding to attain their final correct conformation. A sensitive system exists to prevent misfolded proteins from progressing through the secretory pathway; it directs them toward a degradative pathway [49-51]. The processes that prevent accumulation of unfolded proteins in the ER lumen are regulated by an intracellular signaling pathway known as the Unfolded Protein Response (UPR), which facilitates cellular adaptation to alterations in protein-folding in the ER lumen by expanding the capacity for protein folding. This is accomplished by molecular chaperone proteins (BiP/GRP78). When unfolded proteins accumulate in the ER, BiP/GRP78 releases transmembrane ER proteins (e.g. PERK, IRE1, ATF6) inducing the UPR. In the current study, we explored ER stress genes and proteins in retinas of cbs-/- mice compared to wild type mice. Our data showed an increase in several ER stress genes/ proteins including BiP/GRP78 and PERK. They suggest that ER stress, like oxidative stress and excitotoxicity, is a key mechanism in the retinal neurovasculopathy associated with excess levels of Hcy.
Earlier comprehensive investigation of the retinal vasculature in these mice revealed ischemia, neovascularization and a diminished blood-retinal barrier [33].Accompanying these pathological changes were increased levels of VEGF mRNA and protein. Increased VEGF levels, which we observed also in this study, are associated with increased ER stress [37,38]. Abcouwer and colleagues performed in vitro studies investigating the role of HHcy in upregulating VEGF in ARPE-19 cells via an ER stress-mediated pathway [44], but there have been no investigations of HHcy and ER stress in retina in vivo. The present studies fill that void.
In the current work, we demonstrate upregulation of ER stress genes in the retinas of the cbs-/- mouse, particularly BiP/GRP78 and PERK, providing strong evidence that ER stress is induced in this model. BiP/GRP78 is associated with PERK, which is the major protein responsible for attenuation of mRNA translation during ER stress. It prevents influx of newly synthesized proteins into ER, which is not able to manage the additional protein folding load [52]. However, if the unfolded protein response does not alleviate this stress, the pathways for apoptosis are activated, which includes PERK. Our data show that PERK is elevated at the gene and protein level in the cbs-/- retina. Elevation of PERK can in turn upregulate the pro-apoptotic transcriptional factor CHOP. While we did not observe a significant increase of CHOP at the gene or protein level in retinas of cbs-/- mice by qRT-PCR and western blotting, we did we detect an increase in CHOP in specific retinal layers (nerve fiber layer, ganglion cell inner nuclear layer) using immunohistochemical methods. Since CHOP is implicated in apoptosis, we explored markers of apoptosis and found that both PARP and caspase-3 were elevated significantly in the retinas of severely HHcy mice. Interestingly, we found that the caspase-3 positive cells also labeled positively for the vessel marker isolectin-B4, which may be relevant to the vasculopathy observed in the cbs-/- mice. In summary, the present study presents the first systematic investigation of the role of ER stress in HHcy-induced retinal disease. The evidence suggests that the genes and proteins that are major players in the ER stress pathway are upregulated in retinas of mice with severe HHcy due to a defect in the transsulfuration pathway. The data support a role for ER stress in the pathophysiology associated with HHcy-linked retinal disease. Whether the ER stress observed is causative of the neurovasculopathy or a response to it, is not known. Future studies will explore this and will also investigate whether ER stress plays a role in HHcy-linked retinopathies that are due to deficiencies of other enzymes in the Hcy metabolic pathway, particularly the remethylation pathway. Additional experiments are underway to examine whether inhibiting ER stress has any positive effect on the neurovasculopathy observed in the HHcy-induced retinopathy.
Acknowledgment
The authors thank Yonju Ha, PhD for designing the primers used for analysis of ER stress genes and Mrs. Penny Roon for excellentassistance with histological preparations of mouse retina.
References
- Perła-Kaján J, Twardowski T, Jakubowski H. Mechanisms of homocysteine toxicity in humans. Amino Acids. 2007; 32: 561-572.
- Maron BA, Loscalzo J. The treatment of hyperhomocysteinemia. Annu Rev Med. 2009; 60: 39-54.
- Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ. 2002; 325: 1202.
- Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA. 2002; 288: 2015-2022.
- Agnati LF, Genedani S, Rasio G, Galantucci M, Saltini S, Filaferro M, et al. Studies on homocysteine plasma levels in Alzheimer's patients. Relevance for neurodegeneration. J Neural Transm. 2005; 112: 163-169.
- Hogervorst E, Ribeiro HM, Molyneux A, Budge M, Smith AD. Plasma homocysteine levels, cerebrovascular risk factors, and cerebral white matter changes (leukoaraiosis) in patients with Alzheimer disease. Arch Neurol. 2002; 59: 787-793.
- Semba RD. Retinal Vascular Disease. Nutrition and Health: Handbook of Nutrition and Ophthalmology. Humana Press Inc., Totowa, NJ. 2007; 257-280.
- Wright AD, Martin N, Dodson PM. Homocysteine, folates, and the eye. Eye (Lond). 2008; 22: 989-993.
- Cahill MT, Stinnett SS, Fekrat S. Meta-analysis of plasma homocysteine, serum folate, serum vitamin B(12), and thermolabile MTHFR genotype as risk factors for retinal vascular occlusive disease. Am J Ophthalmol. 2003; 136: 1136-1150.
- Jaksic V, Markovic V, Milenkovic S, Stefanovic I, Jakovic N, Knezevic M, et al. MTHFR C677T homozygous mutation in a patient with pigmentary glaucoma and central retinal vein occlusion. Ophthalmic Res. 2010; 43: 193-196.
- Gao W, Wang YS, Zhang P, Wang HY. Hyperhomocysteinemia and low plasma folate as risk factors for central retinal vein occlusion: a case-control study in a Chinese population. Graefes Arch Clin Exp Ophthalmol. 2006; 244: 1246-1249.
- Gumus K, Kadayifcilar S, Eldem B, Saracbasi O, Ozcebe O, Dundar S, et al. Is elevated level of soluble endothelial protein C receptor a new risk factor for retinal vein occlusion? Clin Experiment Ophthalmol. 2006; 34: 305-311.
- Abu El-Asrar AM, Al-Obeidan SA, Abdel Gader AG. Retinal periphlebitis resembling frosted branch angiitis with nonperfused central retinal vein occlusion. Eur J Ophthalmol. 2003; 13: 807-812.
- Lahey JM, Kearney JJ, Tunc M. Hypercoagulable states and central retinal vein occlusion. Curr Opin Pulm Med. 2003; 9: 385-392.
- Tranchina L, Centofanti M, Oddone F, Tanga L, Roberti G, Liberatoscioli L, et al. Levels of plasma homocysteine in pseudoexfoliation glaucoma. Graefes Arch Clin Exp Ophthalmol. 2011; 249: 443-448.
- Turgut B, Kaya M, Arslan S, Demir T, Güler M, Kaya MK, et al. Levels of circulating homocysteine, vitamin B6, vitamin B12, and folate in different types of open-angle glaucoma. Clin Interv Aging. 2010; 5: 133-139.
- Roedl JB, Bleich S, Reulbach U, Rejdak R, Kornhuber J, Kruse FE, et al. Homocysteine in tear fluid of patients with pseudoexfoliation glaucoma. J Glaucoma. 2007; 16: 234-239.
- Roedl JB, Bleich S, Reulbach U, Rejdak R, Naumann GO, Kruse FE, et al. Vitamin deficiency and hyperhomocysteinemia in pseudoexfoliation glaucoma. J Neural Transm. 2007; 114: 571-575.
- Schlötzer-Schrehardt U. [Oxidative stress and pseudoexfoliation glaucoma]. Klin Monbl Augenheilkd. 2010; 227: 108-113.
- Javadzadeh A, Ghorbanihaghjo A, Bahreini E, Rashtchizadeh N, Argani H, Alizadeh S. Serum paraoxonase phenotype distribution in exudative age-related macular degeneration and its relationship to homocysteine and oxidized low-density lipoprotein. Retina. 2012; 32: 658-666.
- Carrillo-Carrasco N, Venditti CP. Combined methylmalonic acidemia and homocystinuria, cblC type. II. Complications, pathophysiology, and outcomes. J Inherit Metab Dis. 2012; 35: 103-114.
- Javadzadeh A, Ghorbanihaghjo A, Bahreini E, Rashtchizadeh N, Argani H, Alizadeh S, et al. Plasma oxidized LDL and thiol-containing molecules in patients with exudative age-related macular degeneration. Mol Vis. 2010; 16: 2578-2584.
- Lim CP, Loo AV, Khaw KW, Sthaneshwar P, Khang TF, Hassan M, et al. Plasma, aqueous and vitreous homocysteine levels in proliferative diabetic retinopathy. Br J Ophthalmol. 2012; 96: 704-707.
- Cho HC. The Relationship among Homocysteine, Bilirubin, and Diabetic Retinopathy. Diabetes Metab J. 2011; 35: 595-601.
- Satyanarayana A, Balakrishna N, Pitla S, Reddy PY, Mudili S, Lopamudra P, et al. Status of B-vitamins and homocysteine in diabetic retinopathy: association with vitamin-B12 deficiency and hyperhomocysteinemia. PLoS One. 2011; 6: e26747.
- Garovic-Kocic V, Rosenblatt DS. Methionine auxotrophy in inborn errors of cobalamin metabolism. Clin Invest Med. 1992; 15: 395-400.
- Welch GN, Loscalzo J. Homocysteine and atherothrombosis. N Engl J Med. 1998; 338: 1042-1050.
- Maron BA, Loscalzo J. Should hyperhomocysteinemia be treated in patients with atherosclerotic disease? Curr Atheroscler. Rep 2007; 9: 375–383.
- McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969; 56: 111-128.
- Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR, et al. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci U S A. 1995; 92: 1585-1589.
- Ganapathy PS, Moister B, Roon P, Mysona BA, Duplantier J, Dun Y, et al. Endogenous elevation of homocysteine induces retinal neuron death in the cystathionine-beta-synthase mutant mouse. Invest Ophthalmol Vis Sci. 2009; 50: 4460-4470.
- Ganapathy PS, Roon P, Moister TK, Mysona B, Smith SB. Diabetes Accelerates Retinal Neuronal Cell Death In A Mouse Model of Endogenous Hyperhomocysteinemia. Ophthalmol Eye Dis. 2009; 1: 3-11.
- Tawfik A, Al-Shabrawey M, Roon P, Sonne S, Covar JA, Matragoon S. Alterations of retinal vasculature in cystathionine-Beta-synthase mutant mice, a model of hyperhomocysteinemia. Invest Ophthalmol Vis Sci. 2013; 54: 939-949.
- Tawfik A, Markand S, Al-shabrawey M, Henry J, Reynolds J, Bearden SE, et al. Alterations of retinal vasculature in cystathionine-beta-synthase heterozygous mice, a model of mild-moderate hyperhomocysteinemia. Invest Ophthalmol Vis Sci. 2013; 54: 939–949.
- Yu M, Sturgill-Short G, Ganapathy P, Tawfik A, Peachey NS, Smith SB, et al. Age-related changes in visual function in cystathionine-beta-synthase mutant mice, a model of hyperhomocysteinemia. Exp Eye Res. 2012; 96: 124-131.
- Ganapathy PS, White RE, Ha Y, Bozard BR, McNeil PL, Caldwell RW, et al. The role of N-methyl-D-aspartate receptor activation in homocysteine-induced death of retinal ganglion cells. Invest Ophthalmol Vis Sci. 2011; 52: 5515-5524.
- Pereira ER, Frudd K, Awad W, Hendershot LM. Endoplasmic reticulum (ER) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (HIF-1) transcriptional activity on targets like vascular endothelial growth factor (VEGF). J Biol Chem. 2014; 289: 3352-3364.
- Pollreisz A, Afonyushkin T, Oskolkova OV, Gruber F, Bochkov VN, Schmidt-Erfurth U. Retinal pigment epithelium cells produce VEGF in response to oxidized phospholipids through mechanisms involving ATF4 and protein kinase CK2. Exp Eye Res. 2013; 116: 177-184.
- Wang X, Wang G, Kunte M, Shinde V, Gorbatyuk M. Modulation of angiogenesis by genetic manipulation of ATF4 in mouse model of oxygen-induced retinopathy [corrected]. Invest Ophthalmol Vis Sci. 2013; 54: 5995-6002.
- Ha Y, Shanmugam AK, Markand S, Zorrilla E, Ganapathy V, Smith SB, et al. Sigma receptor 1 modulates ER stress and Bcl2 in murine retina. Cell Tissue Res. 2014; 356: 15-27.
- Ha Y, Saul A, Tawfik A, Zorrilla EP, Ganapathy V, Smith SB, et al. Diabetes accelerates retinal ganglion cell dysfunction in mice lacking sigma receptor 1. Mol Vis. 2012; 18: 2860-2870.
- Zhou J, Austin RC. Contributions of hyperhomocysteinemia to atherosclerosis: Causal relationship and potential mechanisms. Biofactors. 2009; 35: 120-129.
- Zhang SX, Sanders E, Fliesler SJ, Wang JJ. Endoplasmic reticulum stress and the unfolded protein responses in retinal degeneration. Exp Eye Res. 2014; 125C: 30-40.
- Roybal CN, Yang S, Sun CW, Hurtado D, Vander Jagt DL, Townes TM, et al. Homocysteine increases the expression of vascular endothelial growth factor by a mechanism involving endoplasmic reticulum stress and transcription factor ATF4. J Biol Chem. 2004; 279: 14844-14852.
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004; 11: 381-389.
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995; 376: 37-43.
- Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995; 81: 801-809.
- Ganapathy PS, Perry RL, Tawfik A, Smith RM, Perry E, Roon P, et al. Homocysteine-mediated modulation of mitochondrial dynamics in retinal ganglion cells. Invest Ophthalmol Vis Sci. 2011; 52: 5551-5558.
- Smith SB. ER stress and retinal disease. Retina (5th Edition). Ryan SJ, Schachat AP, Wilkinson CP, Hinton DR, Sadda S, Wiedemann P, Editors. Elsevier, London 2012.
- Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007; 18: 716-731.
- Oshitari T, Hata N, Yamamoto S. Endoplasmic reticulum stress and diabetic retinopathy. Vasc Health Risk Manag. 2008; 4: 115-122.
- Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013; 1833: 3460-3470.