
Research Article
Austin J Clin Ophthalmol. 2023; 10(2): 1144.
Ligand-Independent Activation of Platelet-Derived Growth Factor Receptor β Promotes Contraction of Retinal Pigment Epithelial Cells
Duan Y1,2#, Wu W3#, Cui J4, Matsubara JA4, Kazlauskas A5, Li X1* and Hetian Lei6*
1Tianjin Key Laboratory of Retinal Functions and Diseases, Tianjin Branch of National Clinical Research Center for Ocular Disease, Eye Institute and School of Optometry, Tianjin Medical University Eye Hospital, China
2Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Tongji Shanxi Hospital, Third Hospital of Shanxi Medical University, China
3Department of Ophthalmology, Hunan Key Laboratory of Ophthalmology, National Clinical Research Center for Geriatric Disorders, Xiangya Hospital of Central South, China
4Department of Ophthalmology and Visual Sciences, The University of British Columbia, Canada
5Department of Ophthalmology, University of Illinois at Chicago, USA
6Shenzhen Eye Hospital, Jinan University, Shenzhen Eye Institute, China
*Corresponding author: Hetian LeiXiaorong Li, Department of Ophthalmology, University of Illinois at Chicago, 251 Fukang Road, Xiqing District, Tianjin 300384, China
Hetian Lei, Shenzhen Eye Hospital, Jinan University, Shenzhen Eye Institute, 18 Zetian Road, Futian District, Shenzhen, China
Received: January 30, 2023; Accepted: March 07, 2023; Published: March 14, 2023
Abstract
Background: Epiretinal membranes in patients with Proliferative Vitreoretinopathy (PVR) consist of extracellular matrix and a number of cell types including Retinal Pigment Epithelial (RPE) cells and fibroblasts, whose contraction causes retinal detachment. In RPE cells depletion of Platelet-Derived Growth Factor (PDGF) receptor (PDGFR) β suppresses vitreous-induced Akt activation, where as in fibroblasts Akt activation through indirect activation of PDGFRa by growth factors outside the PDGF family (non-PDGFs) plays an essential role in experimental PVR. Whether non-PDGFs in the vitreous, however, were also able to activate PDGFRβ in RPE cells remained elusive.
Methods: We showed that expression of a truncated PDGFRβ lacking a PDGF-binding domain in the RPE cells whose PDGFRB gene had been silent using the CRISPR/Cas9 technology restored vitreous-induced Akt activation as well as cell proliferation, epithelial-mesenchymal transition, migration and contraction.
Results: We found that scavenging Reactive Oxygen Species (ROS) with N-acetyl-cysteine and inhibiting Src Family Kinases (SFKs) with their specific inhibitor SU6656 blunted the vitreous-induced activation of the truncated PDGFRβ and Akt as well as the cellular events related to the PVR pathogenesis.
Conclusions: These discoveries suggest that in RPE cells PDGFRβ can be activated indirectly by non-PDGFs in the vitreous via an intracellular pathway of ROS/SFKs to facilitate the development of PVR, thereby providing novel opportunities for PVR therapeutics.
Keywords: Vitreous Indirect Activation; PDGFRβ; Akt; Retinal Pigment Epithelial Cells; Proliferation; Epithelial-Mesenchymal Transition; Migration; Contraction
Introduction
Platelet-Derived Growth Factor (PDGF) receptors (PDGFRs) were in 1994 firstly shown to be expressed in cells within Epi Retinal Membranes (ERMs) from patients with Proliferative Vitreoretinopathy (PVR) [1]. PVR is a fibrotic eye disease, which develops at a rate of 5-10% after surgery correction of a retinal detachment [2-4] and occurs at a rate of 40-60% after open ocular trauma [5,6]. In the PVR pathogenesis retinal cells including Retinal Pigment Epithelial (RPE) cells lodged in the vitreous after retinal repairing surgery or mechanistic retinal damage from Epi-or sub-Retinal Membranes (ERMs) after their proliferation, Epithelial-Mesenchymal Transition (EMT), migration and secretion of extracellular matrix [3,7]. The tracking force of the ERMs causes retinal detachment [8]. At present there is no approved medicine for this eye disease [9]; the only treatment option with the surgery leads to the poor sight recovery [10,11]. Therefore, it is urgent to develop a pharmacological approach for the therapy of PVR.
In the PDGFR family the products of two genes PDGFRA and PDGFRB can form three dimers: PDGFRaa, PDGFRaβ and PDGFRββ, which are Receptor Tyrosine Kinases (RTKs) [12]. There are activated PDGFRa and PDGFRβ in ERMs from PVR patients [13], and in the ERMs there are a variety of cell types including RPE cells, glial cells, fibroblasts and macrophages [8,14]. The PDGF family contains five protein members: PDGF-AA, -BB, -AB, -CC and -DD, which are produced by four genes: PDGFA, PDGFB, PDGFC and PDGFD. Notably, while PDGF-AA is a specific ligand for PDGFRaa, PDGF-BB can bind all the PDGFR dimers: PDGFRaa, -aβ and -β. In addition, PDGF-CC resembling PDGF-AB can bind to PDGFRaa and -aβ, whereas PDGF-DD can bind to PDGFRββ with a strong affinity, but to PDGFRa with a weak affinity [15,16].
Traditionally, binding of a ligand (e.g., PDGF) specifically to a RTK (e.g., PDGFR) induces the RTK’s dimerization and conformation change, leading to activation of its intracellular kinase domain that phosphorylates itself at a number of tyrosine sites. Subsequently, the phosphorylated tyrosine can be bound by intracellular enzymes with a Src Homology (SH)2 domain, for instance, Src, and adaptors including a p85 regulatory subunit of Phosphorinositide 3-Kinases (PI3K) [16-18]. As a consequence, the extracellular signal is transduced to a variety of intracellular enzymatic activation including the signaling pathway of PI3K/Akt. As a serine and threonine kinase, Akt plays an essential role in numerous cellular events including cell growth, survival, dedifferentiation, motility, and metabolism [19,20]. Up-regulation of Akt activity is strongly associated with a number of human diseases including cancer [17,21] and PVR [22-25].
In addition, PDGFRa could be activated indirectly via an intracellular route of Reactive Oxygen Species (ROS) and Src Family Kinases (SFKs) [24,26]. Here, ROS comes from Rac GTPases–regulated Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase [27] and mitochondria due to the decreased autophagy [25]. That is, growth factors outside the PDGF family (non-PDGFs) are able to activate the pathway of PDGFRa/PI3K/Akt/mTORC1 (mammalian target of rapamycin complex 1), resulting in a reduction in autophagy [25]. This module of action has been demonstrated using fibroblasts derived from genetically modified mouse embryos [25,28]; that is, indirect activation of PDGFRa via an intracellular route of ROS/SFKs contributes to the development of PVR [23,29].
RPE cells play a critical role in the PVR pathogenesis because these cells are the major component of ERMs from patients with PVR [8,14]. Thanks to the advent of the technology of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated endonuclease (Cas)9 [6,30-32], we recent discovered that PDGFRβ is the predominant isoform among the PDGFRs in RPEM cells, which were the RPE cells derived from ERMs from patients with PVR [16,33,34], and that PDGFRβ plays an essential role in vitreous-induced activation of Akt in the RPEM cells and cellular events related to the PVR pathogenesis [16]. Thereby, we hypothesized that in RPE cells non-PDGFs in the vitreous were able to activate PDGFRβ indirectly via an intracellular pathway of ROS/SFKs, and a truncated PDGFRβ lacking a PDGF binding domain [35,36] was harnessed to test this hypothesis.
Material and Methods
Major Reagents and Cell Culture
Antibodies against p-Akt (p-S473) (Catalog #: 9271), Akt (Catalog #: 9272), p-PDGFRβ (p-Y751, catalog #: 3166) and PDGFRβ (Catalog #: 3162) were purchased from Cell Signaling Technology (Danvers, MA), antibodies against Ki67 (Catolog #: ab243878) and a-smooth muscle act in (a-SMA) Catalog #: ab5694) were from Abcam (Danvers, MA), a heat shock protein (Hsp)90a (Catalog #: PA3-0137) was from ABR Affinity Bioreagents (Golden, CO) and a β-Actin antibody (Catalog #: sc-47778) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). HRP (Horseradish Peroxidase)-conjugated goat anti-rabbit IgG (Catalog #: sc-2004) and goat anti-mouse IgG (Catalog #: sc-2005) secondary antibodies were ordered from Santa Cruz Biotechnology. Enhanced chemiluminescent substrate for detection of horseradish peroxidase was from Thermo Fisher Scientific (Waltham, MA). N-Acetyl-Cysteine (NAC, a scavenger of ROS) and SU6656 (a selective inhibitor of Src family kinases) [23] were purchased from Sigma (St. Luis, MO) and Calbiochem (San Diego, CA), respectively.
Rabbit Vitreous (RV) was prepared by dissection from the rabbit eyeball when it was still frozen, and the thawed vitreous was centrifuged at 4°C for 5 minutes at 10,000×g. The resulting supernatant was used for all analyses. There is hardly detectable PDGF in RV as demonstrated previously [23].
RPEM cells were derived from an epiretinal membrane from a patient with grade C PVR and expressed RPE-cells’ markers including keratin as described previously [33; Xin, 2020 #1365]. These RPEM cells were gifts from Dr. Joanne Matsubara at the University of British Columbia, Canada, and grown in Dulbecco’s Modified Eagle’s Medium/nutrient mixture (DMEM/F12, Thermo Fisher Scientific) supplemented with 10% Fetal Bovine Serum (FBS) and antibiotics of streptomycin (50ug/ml) and penicillin (50 units/ml). When these cells were sub-cultured, 1:2 split was performed in their 90% confluence.
Human Embryonic Kidney (HEK) 293GPG cells were gifts from the Kazlauskas lab at the Schepens Eye Research Institute (Boston, MA) and were grown in high-glucose (4.5g/L) DMEM supplemented with 10% FBS, G418 (0.3mg/ml), tetracycline (1ug/ml) and puromycin (2ug/ml). These 293GPG cells were stably transfected with genes of vesicular stomatitis virus including the gag, pol, and the VSV-Ggene. All mammalian cells were cultured at 37°C in a humidified incubator with 5% CO2 [37].
Construction of PDGFRβΔx
Construction of PDGFRβΔx was completed in two steps. First, the PDGFRβΔx lacking amino acids 38 to 442 of the human PDGFRβwas cloned into the PVZ-ApaI-NotI-EcoRI-XbaI-SalI-PstI-HindII vector [35] using EcoRI/Xbal. Then the digested PDGFRβΔxDNA fragment with EcoRI/SalI was subcloned into pLXSHD-EcoRI-HpaI-XhoI-BamHI vector digested withEcoRI/Xhol. The resultant construct was termed pLXSHD-PDGFRβΔx and verified by Sanger DNA sequencing at the MGH DNA core facility (Cambridge, MA).
Production of Retrovirus
In the production of retrovirus there were three steps: transfection, collection of retrovirus, and concentration. Transfection: Gently mix lipofectamine 2000 (156μl, Thermo Fisher Scientific) pLXSHDor pLXSHD-PDGFRβΔx (25μg) in an OPTIMEM medium (1.8ml, Thermo Fisher Scientific), incubate these mixtures at room temperature for 30 minutes so as to form liposomesentraping the DNA, and then transfer these liposomes drop wise into the 293GPG cells, which were in about 70% confluence in a 15-cm cell culture dish. Notably, during transfection, the growth medium was changed to 10ml OPTIMEM, and after transfection for 7-10 hours, a 12ml virus-producing medium (high glucose DMEM with 10% FBS) was added. In the following morning, a 20ml fresh virus-producing medium was used to replace the old medium. Collection of retrovirus: harvest the culture media containing retroviruses after transfection at 48,72,96,120 hours, and spin at 1500rpm for 10 minutes to remove cells and debris. Concentration: Spin the supernatant containing the virus at 25,000×g, 4°C for 90 minutes, dissolve the white pellet on the bottom of the centrifuge tube in 300μl of sterile TNE buffer (50mM Tris pH7.8, 130mM NaCl, 1mM EDTA), and then gently rotate the solution overnight at 4°C to obtain the retrovirus [38].
Expression of PDGFRβΔx in RPEM cells
Depletion of PDGFRβ in RPEM cells was achieved using the CRISPR/Cas9 technology with a PB3 sgRNA (GCCTGGTCGTCACACCCCC) guiding SpCas9 to cleave human genomic PDGFRB at exon 3 as described previously [16]. These PDGFRβ-depleted RPEM cells were infected by the concentrated retrovirus in DMEM supplemented with 10% FBS and 8μg/mL polybrene (hexadimethrine bromide; Sigma, St. Louis, MO). Cells expressing PDGFRβΔx were selected in a histidine-free DMEM supplemented with 2mM L-histidinol dihydrochoride (Sigma), and the levels of the PDGFRβΔx in these cells were determined by western blot with an anti–PDGFRβ antibody recognizing the intracellular domain of PDGFRβ [16].
Western Blot
As described previously [16], proteins in cell lysates after centrifugal clarification at 13,000×g for 10 minutes were mixed with a sample buffer and denatured by boiling for 5 minutes. Subsequently the soluble proteins in the sample buffer were separated by 10% SDS-polyacrylamide gel electrophoresis. The proteins in the gel were then transferred to polyvinylidene difluoride membranes for western blot analysis with desired antibodies [16].
Cell Proliferation Assay
RPEM cells after trypsin detachment were counted and seeded into wells of a 24-well plate in DMEM/F12 with 10% FBS at a density of 3×104 cells/well. After attaching the plate, the RPEM cells were treated with DMEM/F12 only or RV (1:3 dilution in DMEM/F12) with additional NAC (5mM), SU6656 (1μM) or their solvent. The cells were trypsin detached after treatment for 48 hours for cell counting. Each experimental condition was treated in duplicate, and data from three independent experiments were subjected to statistic analysis [26,29].
In addition, after reaching 80% confluence, cells were cultured in a serum-free medium overnight, and then the cells were treated with RV plus Ki67 for additional 48 hours. Next, a Ki67 primary antibody was incubated with the cells overnight at 4°C, and the fluorescent secondary antibody was incubated for 1 hour in the dark at room temperature. Finally, the cells were stained with 4’,6-Diamidino-2-Phenylindole (DAPI) for 10 minutes, and the slides were mounted for photographs. The data were analyzed in Image J software for counting the Ki67-stained cell numbers [39].
An Epithelial-Mesenchymal Transition Assay
When RPEM cells grew to 90% confluence in 24-well culture plates, they were serum-starved overnight and then treated with RV or RV in the presence of NAC, SU6656 for additional 48 hours. The resultant lysates were subjected to western blot using desired antibodies, among which there was an antibody against a-SMA, a protein marker of EMT [40,41].
A Scratch Wound Assay
When RPEM cells grew to near confluence in wells of a 24-well plate, the wells were scratched with a 200μl pipet tip. After washing with Phosphate-Buffered Solution (PBS), the cells were treated with a medium of DMEM/F12 only or RV (1:3 dilution in DMEM/F12) with or without addition of NAC (5mM) or SU6656 (1μM). 16 hours later the wound areas were photographed and analyzed with Adobe Photoshop CS6 software. Data from three independent experiments were subjected to statistic analysis [16,42].
Contraction Assay
RPEM cells were trypsin detached, counted and mixed with collagen I (INAMED, Fremont, CA) on ice. The final collagen I concentration was 1.5mg/ml, cell density was at 1×106cells/ml, and the pH value was adjusted to 7.2 as described previously [23,29]. 300μl cell-gel mixture was transferred into wells of a 24-well plate, which had been preincubated with 5mg/ml bovine serum albumin/PBS for at least 8 hours. 90 minutes later the collagen gel was polymerized at 37°C, and 0.5ml DMEM/F12 or RV (1:3 dilution in DMEM/F12) with or without additional NAC (5mM) or SU6656 (1μM). The gel diameter was measured and photographed on day 2 or 3 for further analysis. Data from three independent experiments were subjected to statistic analysis [16,29,43].
Statistics
As described previously [8,16], data were collected from three independent experiments for analysis using ordinary one-way analysis of variance (ANOVA) followed by Tukey’s Honest Significant Difference (HSD) post hoc test. A significant difference between groups was determined by a P value less than 0.05.
Institutional Review Board Statement
Collection of epiretinal membranes from patients with proliferative vitreoretinopathy for this study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board of the Vancouver Hospital and University of British Columbia Clinical Research Ethics Board.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Animal Use
The protocol for the use of animals was approved by the Schepens Eye Research Institute Animal Care and Use Committee (Boston, MA), and all animal surgeries adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and followed the ARRIVE guidelines (https://arriveguidelines.org).
Results
PDGFRβ in the RPE cells plays a critical role in vitreous-induced activation of Akt and cellular responses intrinsic to the pathogenesis of PVR [16], but whether non-PDGFs in the vitreous play a part in these signaling and cellular events was unknown. Our goal herein was to answer this intriguing question, and our hypothesis was that non-PDGFs were able to indirectly activate PDGFRβ via an intracellular pathway of ROS/SFKs based previous findings [26]. To test this hypothesis, we employed a truncated PDGFRβ lacking a PDGF binding domain [35,36] and specific inhibitors of ROS and SFKs. The experiments showed that non-PDGFs in the vitreous could activate PDGFRβ and Akt via the intracellular domain of PDGFR, leading to cellular responses of proliferation, EMT, migration and contraction related to the development of PVR.
Expression of a Truncated PDGFRβ Lacking a PDGF Binding Domain in the PDGFRB-Silent RPE Cells Restores Vitreous-Induced Akt Activation
To seek an answer to the question whether non-PDGFs in the vitreous activated PDGFRβ via an intracellular pathway in the RPE cells, a truncated PDGFRβ that did not have a PDGF-binding domain that spans from 38th to 442th amino acids of PDGFRβ [35] was expressed in the RPE cells whose PDGFRB had been silent using the CRISPR/Cas9 technology [16]. The single guide (sg) RNA denoted as PB3 guiding SpCas9 to specifically edit genomic PDGFRB could not recognize the mutant PDGFRB encoding the truncated PDGFRβ (Figure 1). In this CRISPR/Cas9 system, the PB3-sgRNA was designed to target human PDGFRB at exon 3, and the three nucleotides (GGG) of the Protospacer Adjacent Motif (PAM) for the PB3-sgRNA were absent from the mutant PDGFRB (Figure 1A). Thereby we constructed a retroviral vector with the PDGFRB mutant to express the truncated PDGFRβ. This truncated PDGFRβ was named as PDGFRβx (Figure 1B). The retrovirus containing DNA sequence coding PDGFRβ△x was used to infect the PDGFRB-silent RPEM cells (Figure 2). As expected, PDGFRβ△x was successfully expressed in the PDGFRβ-depleted RPEM cells, and PDGF-BB activated PDGFRβ and Akt in the control RPEM cells, but neither in the PDGFRB-silent or in the PDGFRβ△x-expressed RPEM cells (Figure 2). However, RV, in which there are no detectable PDGFs as determined previously [26], activated not only PDGFRβ but also PDGFRβ△x in the PDGFRβ or PDGFRβ△x-expressed RPEM cells (Figure 2). Notably, expression of PDGFRβ△x in the PDGFRβ-depleted RPEM cells restored vitreous-induced activation of Akt (Figure 2).
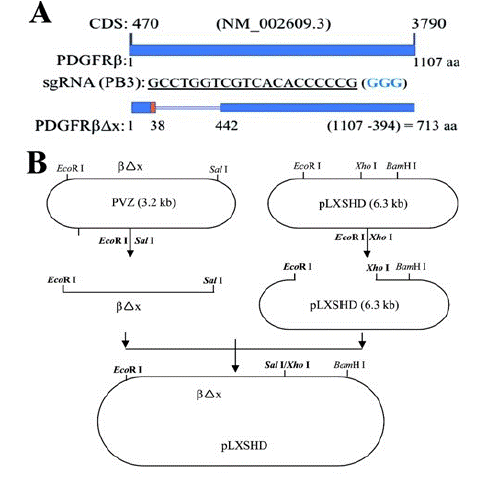
Figure 1: Construction of a retroviral vector to express a truncated PDGFRβ.
A. The Coding Sequence (CDS) of human PDGFRB from nucleotide 470 to nucleotide 3790 (NM_002609.3) codes human PDGFR: 1107 Amino Acids (aa) [48]. The protospacer GCCTGGTCGTCACACCCCCG (563-583) from human PDGFRB exon 3 was used to generate a specific guide RNA (PB3-sgRNA), which guided Streptococcus Pyogenes (Sp) Cas9 to cleave human genomic PDGFRB in RPE cells, resulting in depletion of PDGFRβ. The blue colored three nucleotides GGG is Protospacer Adjacent Motif (PAM) for SpCas9 recognition [16]. The truncated human PDGFRβ lacking the 38th-442th amino acids of its wild type PDGFRβ was denoted as PDGFRβΔx and its coding sequence was not targeted by PB3-sgRNA.
B. The coding sequence of PDGFRβΔx was subcloned from a PVZ vector [35] to a pLXSHD retrovial vector.

Figure 2: Vitreous induces Akt activation via the truncated PDGFRβ.
A. Representative images of western blot with indicated antibodies. When RPEM cells grew to 80% confluence, they were serum-starved overnight and then treated with RV, PDGF-B or RV in the presence of NAC or SU6656 for 15 minutes. The resultant lysates were subjected to western blot using indicated antibodies. RV (Rabbit Vitreous) diluted (1:3) in the medium of DMEM/F12, PDGF-B (10ng/ml), NAC: N-acetyl-cysteine (5mM), SU: SU6656 (1μM), a specific inhibitor of SFKs. EV: empty vector (pLXSHD), βΔx: a truncated PDGFRβ lacking the PDGF-binding domain.
B. The intensity of the p-Akt bands in A was first normalized to that of the corresponding β-Actin bands and then calculated to establish the ratio of the control in the first lane, shown as “Fold”. The mean ± Standard Deviation (SD) of the three independent experiments is shown; *** denotes p<0.001, and NS stands for not significant using ANOVA.
To investigate whether ROS-mediated Src Family Kinases (SFKs) played a part in RV (non-PDGFs) -induced activation of PDGFRβ in RPEM cells as they do for RV-induced PDGFRa activation in fibroblasts [26], we treated the PDGFRβ△x-expressed RPEM cells with RV in the presence of either a ROS scavenger N-Acetyl-Cysteine (NAC) or a SFKs-specific inhibitor (SU6656). Previous experiments identified that 5 mM NAC or 1μM SU6656 could effectively inhibit vitreous-induced Akt activation and PVR-related cellular events without obvious toxicity to RPE cells [23,25; Blake, 2000 #64, 26]. As predicted, Western blot analysis showed that both 5mM NAC and 1μM SU6656 inhibited RV-induced activation of PDGFRβ△x and Akt (Figure 2), suggesting that both ROS and SFKs are required for non-PDGFs-induced activation of PDGFRβin RPEM cells.
Expression of PDGFRβ△x in the PDGFRβ-Depleted RPE Cells Recovers Vitreous-Induced Cell Proliferation, EMT, and Migration
Akt activation is essential for a great many of cellular responses including cell proliferation, EMT, and migration [19], which are intrinsic to the PVR pathogenesis [8,16]. While vitreous stimulation could induce proliferation and migration of RPEM cells [8,16], PDGFRβ absence from RPEM cells significantly dampens vitreous-induced cellular events [16]. To this end, we investigated if PDGFRβ△x expression in the PDGFRB-silent RPEM cells could resume vitreous-induced cell proliferation and migration. As shown in (Figure 3), while PDGF-BB stimulated RPEM cell’s proliferation, but it failed to do so in those cells expressing PDGFRβ△x. Nevertheless, vitreous stimulated more proliferation of RPEM cells expressing PDGFRβ△x than that of those PDGFRB-silent cells as shown in (Figure 3 and Supplemental Figure 1). Furthermore, both the ROS scavenger NAC and the SFKs’ inhibitor SU6656 hindered the vitreous-induced cell proliferation of the RPEM cells with PDGFRβ△x.
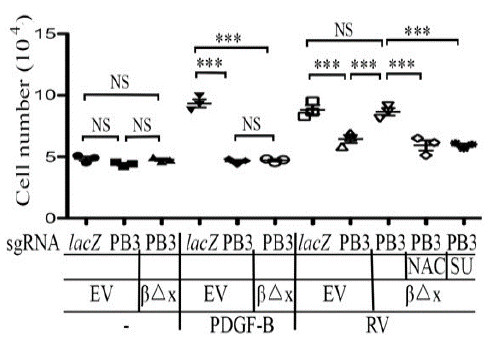
Figure 3: Vitreous stimulates cell proliferation via the truncated PDGFRβ.
RPEM cells characterized in Figure 2 were treated with DMEM/F12, PDGF-B (20ng/ml) or RV diluted (1:3) in DMEM/F12 in the presence or absence of NAC (5mM), or SU: (SU6656, 1μM). After treatment for 48 hours the cells were counted with a hemocytometer. The mean ± SD of three independent experiments is shown; *** denotes p< 0.001, and NS stands for not significant using ANOVA.
EMT is a critical step in the development of PVR [40]. We next evaluated if suppression of PDGFRβ could blunt RV-induced expression of a-SMA, a protein marker of EMT. As shown in (Figure 4), while RV boosted a-SMA expression, depletion of PDGFRβ in RPEM cells lessened a-SMA expression, where as introduction of PDGFRβ△x into these cells restored a-SMA expression induced by RV. Similar to the prior findings, both NAC and SU6656 blocked RV-induced expression of a-SMA in the RPEM cells with PDGFRβ△x (Figure 4), suggesting ligand-independent activation of PDGFR also plays an important part in RV-induced EMT.
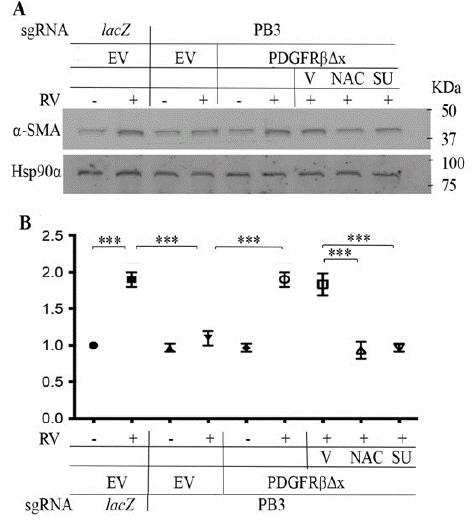
Figure 4: Vitreous stimulates EMT via the truncated PDGFRβ.
A. Representative images of western blot with indicated antibodies. a-SMA is a protein marker of EMT, and Hsp90a is a loading control. RV diluted (1:3) in DMEM/F12, NAC: N-acetyl-cysteine (5mM), SU: SU6656 (1μM). EV: Empty Vector, βΔx: a truncated PDGFRβ lacking the PDGF-binding domain.
B. The intensity of a-SMA bands in A was first normalized to that of the corresponding Hsp90a bands and then calculated to establish the ratio of the control in the first lane, shown as “Fold”. The mean ± Standard Deviation (SD) of the three independent experiments is shown; *** denotes p<0.001 using ANOVA (an ordinary one-way analysis of variance).
We next employed a scratch wound assay to estimate the capability of cell migration as previously described [8,16,42]. As shown in (Figure 5), PDGF-BB enhanced migration of RPEM cells; however, it did not, but vitreous did, induce more migration of the RPEM cells expressing PDGFRβ△x than that of those PDGFRB-silent cells. As expected, both NAC and SU6656 impeded vitreous-induced migration of PDGFRβ△x-expressed RPEM cells (Figure 5).
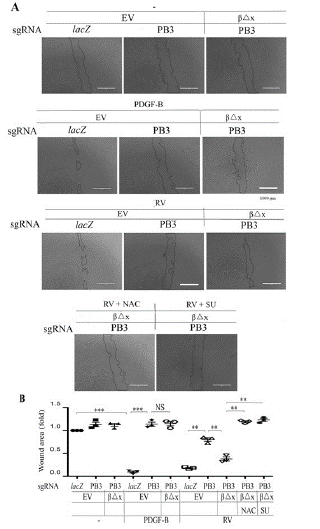
Figure 5: Vitreous stimulates cell migration via the truncated PDGFRβ.
A. Confluent RPEM cells were subjected to a scratch wound healing assay. PDGF-B: 20ng/ml, RVdiluted (1:3) in DMEM/F12, NAC: 5mM, or SU (SU6656, 1μM). Representative images of cell migration shown were taken after 16 hours. Scale bar: 1000μm.
B. Shown is the mean ± SD of three independent experiments. *** denotes p<0.001, and NS stands for not significant using ANOVA.
Expression of PDGFRβ△x in the PDGFRβ-Depleted RPE Cells Restores Vitreous-Induced Cell Contraction
In the pathogenesis of PVR, ERMs consisting of extracellular matrix and cells including RPE cells contract, resulting in the retinal detachment [8,14]. To investigate the molecular mechanism of the PVR pathogenesis, an in vitro assay with collagen and RPEMs was established to mimic the in vivo contraction process. In this assay, the characterized RPEM cells in (Figure 2) were firstly mixed with collagen I to form a cell-collagen mixture, analogy to the ERMs. Subsequently, a DMEM/F12 medium with agents (PDGF-BB or RV) in the presence or absence of NAC (5mM) or SU6656 (1μM) was added onto the collagen gel. This procedure was to mimic the in vivo environment with a drug treatment.
As shown in (Figure 6), PDGF-BB induced a collagen-gel contraction of the RPEM cells expressing PDGFRβ, but it failed to do so in either PDGFRB-silent or PDGFRβ△x-expressing RPEM cells. In addition, depletion of PDGFRβ suppressed vitreous-stimulated contraction of RPEM cells, where as expression of PDGFRβ△x resumed the contractile capability of the engineered RPEM cells (Figure 6). Furthermore, both NAC (5mM) and SU6656 (1M) blocked vitreous-induced contraction of the PDGFRβ△x-expressing RPEM cells, demonstrating that expression of PDGFRβ△x in the PDGFRB-silent RPEM cells restored the vitreous-induced cell contraction (Figure 6), suggesting that vitreal transactivation of PDGFRβ via an intracellular pathway of ROS/SFKs plays a crucial part in PVR-related cellular events.
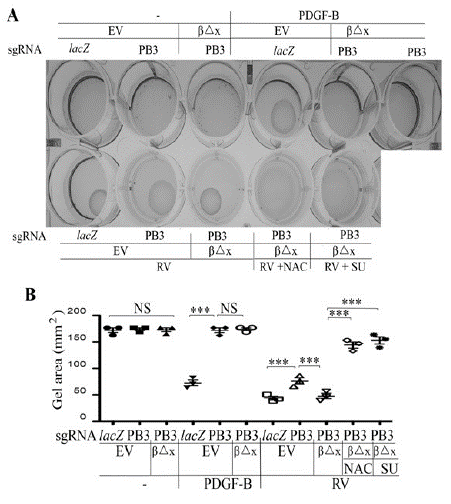
Figure 6: Vitreous induces cell contraction via the truncated PDGFRβ.
A. The defined RPEM cells were subjected to a collagen contraction assay. DMEM/F12 alone (-), PDGF-B (20ng/ml) or RV diluted (1:3) in DMEM/F12, NAC: 5mM, and SU: (SU6656, 1μM). Shown are images from representative of three independent experiments.
B. Shown is the mean ± SD of the three independent experiments. *** denotes p<0.001, and NS indicates not significant using ANOVA.
Discussion
In this report we have showed that in RPE cells from epiretinal membranes (RPEM cells) activation of PDGFRβ by non-PDGFs in the vitreous via the ROS/SFKs pathway is critical for the PVR-related signaling events (e.g., Akt activation) (Figure 2) and cellular responses (e.g., proliferation, EMT, migration and contraction) (Figures 3-6). In the extracellular domain of PDGFRβ there are 5-Ig-like subdomains (D1-D5). Ligand binding is limited to the D2 and D3 domains, which are missing in the truncated PDGFRβ that we employed to test our hypothesis in this report. Notably, D1 is a small domain containing three N-linked glycans, and the membrane proximal D4 and D5 domains play an important part in the PDGFRβ activation [36].
We previously found that in fibroblasts vitreal activation of PDGFRβ intracellularly is hindered by high expression of RasGAP [37], and that in RPE cells expressional levels of RasGAP are lower than those in fibroblasts so that PDGFRβ could play a central role in patient vitreous-stimulated contraction of RPEM cells [16]. In these RPE cells PDGFRa expression is very low so that Akt activation could hardly be induced by PDGF-AA, a PDGFRa-specific ligand [16]. In addition, in RV there is no detectable PDGF [26]; thereby we assume that in the PVR pathogenesis the dislocated RPE cells appear in the foreign environment of the normal vitreous in which there are growth factors outside the PDGF family (non-PDGFs) that are able to activate PDGFRβ. This mechanism of PDGFRβ activation in RPE cells is similar to that PDGFRa is activated by non-PDGFs in fibroblasts via the intracellular pathway of ROS/SFKs [23, 25, 26]. In fact, we revealed that both neutralizing ROS by NAC and inhibiting SFKs by SU6656 in RPEM cells attenuated vitreous-stimulating Akt activation as well as cell proliferation, EMT, migration and contraction (Figures 2-6). Thereby, we propose that non-PDGFs in the vitreous binding to their own respective receptors activate them, resulting in an increase in intracellular levels of ROS, which activate SFKs, which subsequently somehow activate PDGFRβ. This module of intracellular activation of PDGFRβ is also able to engage its downstream signaling events and cellular responses including proliferation, migration, and contraction (Figure 7). These extra signaling events and cellular responses are closely linked with the pathogenesis of PVR (Figure 7).

Figure 7: Schematic diagram of non-PDGFs indirectly activating PDGFRβand its downstream signaling pathway of PI3K/Akt to promote cellular responses intrinsic to PVR pathogenesis.
Non-PDGFs (growth factors outside the PDGF family in the vitreous) activate their own receptors, stimulating production of intracellular reactive oxygen species (ROS) that activates Src Family Kinases (SFKs), which activate PDGFRβ. As a result, the PI3K/Akt signaling is enhanced in Retinal Pigment Epithelial (RPE) cells for promotion of cellular responses including cell proliferation, migration and contraction, leading to the pathogenesis related to Proliferative Vitreoretinopathy (PVR).
In the ERMs there are four major cell types: RPE cells, glia cells, fibroblasts and macrophages [44,45]. The findings reported herein that in RPEM cells PDGFRβ is activated indirectly by non-PDGFs in the vitreous, together with our previous discoveries that PDGFRa is activated indirectly in fibroblasts [24-26] place the PDGFR family into the frontier for preventing PVR [1]. The novelty of this report is that not only could the traditional PDGF binding activate PDGFRβ in RPE cells, but also non-PDGFs in the vitreous could indirectly activate PDGFRβ through intracellular mechanisms in which ROS and SFKs play an important part (Figures 2-6). As a matter of fact, indirect activation of PDGFRβ has been reported in other disease models [35,46,47].
Notably, the reason we herein used normal Rabbit Vitreous (RV) instead of human vitreous was because in the RV there is hardly detectable PDGF, whereas in the clinic human vitreous from patients with PVR there are rich PDGFs including PDGF-BB [3], which can activate PDGFRβ, so we cannot distinguish the modes of direct (ligand binding) and indirect (non-PDGFs) activation by using patient vitreoous. Hence, the RV is a very good source of non-PDGFs, facilitating us to determine if non-PDGFs could activate PDGFRβ indirectly. Nevertheless, there are certain limitations in using RV instead of human vitreous because RV is different from human vitreous and our ultimate goal is to prevent humans from PVR. In addition, the RPEM cells we used in this research were derived from a single ERM from a patient with grade C PVR; thereby our present investigation also has a certain limitation as there may be heterogeneity among the cells in ERMs from different patients with PVR.
Conclusion
Our discoveries reported here in improve our understanding of the mechanism by which PDGFRβ can be activated by non-PDGFs in the vitreous via an intracellular route of ROS/SFKs and provide a conceptual foundation for preventing PVR by inhibiting PDGFR transactivation (ligand-independent activation).
Acknowledgements
YD performed most of the experiments and analyzed the results; XL conceived experiments and revised the manuscript, and HL designed experiments, analyzed data and wrote the manuscript.
Funding
This research was supported by Nature Science Foundation of Shanxi province (202103021224345), Health Commission of Shanxi Province (2020004), Research Project Supported by Shanxi Scholarship Council of China (2022-203), Shanxi Bethune Hospital Foundation (2021RC005), and Shanxi Bethune Hospital Education and Teaching Reform Foundation (2022Jx22) to YD, by National Natural Science Foundation of China (82171085) and Natural Science Foundation of Tianjin (19JCZDJC64000) to XL, by National Natural Science Foundation of China (82070989) to HL, and by Sanming Project of Medicine in Shenzhen (SZSM201812090) to HL. No funding bodies had any role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
All the authors declare that there is no conflict interest in this work.
References
- Robbins SG, Mixon RN, Wilson DJ, Hart CE, Robertson JE, et al. Platelet-derived growth factor ligands and receptors immunolocalized in proliferative retinal diseases. Invest Ophthalmol Vis Sci. 1994; 35: 3649-3663.
- Charteris DG. Proliferative vitreoretinopathy: pathobiology, surgical management, and adjunctive treatment. Br J Ophthalmol. 1995; 79: 953-960.
- Pennock S, Haddock LJ, Eliott D, Mukai S, Kazlauskas A. Is neutralizing vitreal growth factors a viable strategy to prevent proliferative vitreoretinopathy?. Prog Retin Eye Res. 2014; 40: 16-34.
- Schiff L, Boles NC, Fernandes M, Nachmani B, Gentile R, et al. P38 inhibition reverses TGFbeta1 and TNFalpha-induced contraction in a model of proliferative vitreoretinopathy. Commun Biol. 2019; 2: 162.
- Abdullatif AM, Macky TA, Abdullatif MM, Nassar K, Grisanti S, et al. Intravitreal decorin preventing proliferative vitreoretinopathy in perforating injuries: a pilot study. Graefes Arch Clin Exp Ophthalmol. 2018; 256: 2473-2481.
- Blackford BG, Justin GA, Baker KM, Brooks DI, Wang HH, et al. Proliferative Vitreoretinopathy After Combat Ocular Trauma in Operation Iraqi Freedom and Operation Enduring Freedom: 2001-2011. Ophthalmic Surg Lasers Imaging Retina. 2020; 51: 556-563.
- Zhang Y, Wang K, Pan J, Yang S, Yao H, et al. Exosomes mediate an epithelial-mesenchymal transition cascade in retinal pigment epithelial cells: Implications for proliferative vitreoretinopathy. J Cell Mol Med. 2020; 24: 13324-13335.
- Xin T, Han H, Wu W, Huang X, Cui J, et al. Idelalisib inhibits vitreous-induced Akt activation and proliferation of retinal pigment epithelial cells from epiretinal membranes. Exp Eye Res. 2020; 190: 107884.
- Guber J, Lang C, Scholl HPN, Guber I, Valmaggia C. Successful Treatment of Peripheral Proliferative Vitreoretinopathy with Cryocoagulation During Retinal Detachment Repair - A New Surgical Technique. Clin Ophthalmol. 2020; 14: 1413-1416.
- Xiao Y, Choi KS, Warther D, Huffman K, Landeros S, et al. A sustained dual drug delivery system for proliferative vitreoretinopathy. Drug Deliv. 2020; 27: 1461-1473.
- Chaudhary R, Scott RAH, Wallace G, Berry M, Logan A, et al. Inflammatory and Fibrogenic Factors in Proliferative Vitreoretinopathy Development. Transl Vis Sci Technol. 2020; 9: 23.
- Kazlauskas A. PDGFs and their receptors. Gene. 2017; 614: 1-7.
- Cui J, Lei H, Samad A, Basavanthappa S, Maberley D, et al. PDGF receptors are activated in human epiretinal membranes. Exp Eye Res. 2009; 88: 438-444.
- Campochiaro PA. Pathogenic mechanisms in proliferative vitreoretinopathy. Arch Ophthalmol. 1997; 115: 237-241.
- Kumar A, Li X. PDGF-C and PDGF-D in ocular diseases. Mol Aspects Med. 2018; 62: 33-43.
- Yang Y, Huang X, Ma G, Cui J, Matsubara JA, et al. PDGFRbeta plays an essential role in patient vitreous-stimulated contraction of retinal pigment epithelial cells from epiretinal membranes. Exp Eye Res. 2020; 197: 108116.
- Lee KM, Guerrero-Zotano AL, Servetto A, Sudhan DR, Lin CC, et al. Proline rich 11 (PRR11) overexpression amplifies PI3K signaling and promotes antiestrogen resistance in breast cancer. Nat Commun. 2020; 11: 5488.
- Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, et al. The PI3K Pathway in Human Disease. Cell. 2017; 170: 605-635.
- Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell. 2017; 169: 381-405.
- Bilanges B, Posor Y, Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol. 2019; 20: 515-534.
- Goncalves MD, Hopkins BD, Cantley LC. Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N Engl J Med. 2018; 379: 2052-2062.
- Ikuno Y, Leong FL, Kazlauskas A. PI3K and PLCgamma play a central role in experimental PVR. Invest Ophthalmol Vis Sci. 2002; 43: 483-489.
- Lei H, Velez G, Hovland P, Hirose T, Gilbertson D, et al. Growth factors outside the PDGF family drive experimental PVR. Invest Ophthalmol Vis Sci. 2009; 50: 3394-3403.
- Lei H, Velez G, Kazlauskas A. Pathological signaling via platelet-derived growth factor receptor {alpha} involves chronic activation of Akt and suppression of p53. Mol Cell Biol. 2011; 31: 1788-1799.
- Lei H, Kazlauskas A. A reactive oxygen species-mediated, self-perpetuating loop persistently activates platelet-derived growth factor receptor alpha. Mol Cell Biol. 2014; 34: 110-122.
- Lei H, Kazlauskas A. Growth factors outside of the PDGF family employ ROS/SFKs to activate PDGF receptor alpha and thereby promote proliferation and survival of cells. J Biol Chem. 2009; 284: 6329-6336.
- Dieudonne SC, La Heij EC, Diederen R, Kessels AG, Liem AT, et al. High TGF-beta2 levels during primary retinal detachment may protect against proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2004; 45: 4113-4118.
- Andrews A, Balciunaite E, Leong FL, Tallquist M, Soriano P, et al. Platelet-derived growth factor plays a key role in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1999; 40: 2683-2689.
- Lei H, Velez G, Cui J, Samad A, Maberley D, et al. N-Acetylcysteine Suppresses Retinal Detachment in an Experimental Model of Proliferative Vitreoretinopathy. Am J Pathol. 2010; 177: 132-140.
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337: 816-821.
- Huang X, Zhou G, Wu W, Duan Y, Ma G, et al. Genome editing abrogates angiogenesis in vivo. Nat Commun. 2017; 8: 112.
- Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020; 578: 229-236.
- Wong CA, Potter MJ, Cui JZ, Chang TS, Ma P, Maberley AL, et al. Induction of proliferative vitreoretinopathy by a unique line of human retinal pigment epithelial cells. Can J Ophthalmol. 2002; 37: 211-220.
- Zhang H, Shang Q, An J, Wang C, Ma J. Crocetin inhibits PDGF-BB-induced proliferation and migration of retinal pigment epithelial cells. Eur J Pharmacol. 2019; 842: 329-337.
- Drummond-Barbosa DA, Vaillancourt RR, Kazlauskas A, DiMaio D. Ligand-independent activation of the platelet-derived growth factor beta receptor: requirements for bovine papillomavirus E5-induced mitogenic signaling. Mol Cell Biol. 1995; 15: 2570-2581.
- Shim AH, Liu H, Focia PJ, Chen X, Lin PC, et al. Structures of a platelet-derived growth factor/propeptide complex and a platelet-derived growth factor/receptor complex. Proc Natl Acad Sci USA. 2010; 107: 11307-11312.
- Lei H, Qian CX, Lei J, Haddock LJ, Mukai S, et al. RasGAP Promotes Autophagy and Thereby Suppresses Platelet-Derived Growth Factor Receptor-Mediated Signaling Events, Cellular Responses, and Pathology. Mol Cell Biol. 2015; 35: 1673-1685.
- Lei H, Romeo G, Kazlauskas A. Heat shock protein 90alpha-dependent translocation of annexin II to the surface of endothelial cells modulates plasmin activity in the diabetic rat aorta. Circ Res. 2004; 94: 902-909.
- Wu W, Xu H, Meng Z, Zhu J, Xiong S, et al. Axl Is Essential for in-vitro Angiogenesis Induced by Vitreous From Patients With Proliferative Diabetic Retinopathy. Front Med (Lausanne). 2021; 8: 787150.
- Yang S, Li H, Yao H, Zhang Y, Bao H, et al. Long noncoding RNA ERLR mediates epithelial-mesenchymal transition of retinal pigment epithelial cells and promotes experimental proliferative vitreoretinopathy. Cell Death Differ. 2021; 28: 2351-2366.
- Liu B SJ, Han H, Hu Z, Chen N, Cui J, et al. Blockade of MDM2 with inactive Cas9 prevents epithelial to mesenchymal transition in retinal pigment epithelial cells. Lab Invest. 2019; 99: 1874-1886.
- Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007; 2: 329-333.
- Lei H, Rheaume MA, Velez G, Mukai S, Kazlauskas A. Expression of PDGFR{alpha} Is a Determinant of the PVR Potential of ARPE19 Cells. Invest Ophthalmol Vis Sci. 2011; 52: 5016-5021.
- Hinton DR, He S, Jin ML, Barron E, Ryan SJ. Novel growth factors involved in the pathogenesis of proliferative vitreoretinopathy. Eye (Lond). 2002; 16: 422-428.
- Agrawal RN, He S, Spee C, Cui JZ, Ryan SJ, et al. In vivo models of proliferative vitreoretinopathy. Nat Protoc. 2007; 2: 67-77.
- Herrlich A, Daub H, Knebel A, Herrlich P, Ullrich A, et al. Ligand-independent activation of platelet-derived growth factor receptor is a necessary intermediate in lysophosphatidic, acid-stimulated mitogenic activity in L cells. Proc Natl Acad Sci USA. 1998; 95: 8985-8990.
- Liu Y, Li M, Warburton RR, Hill NS, Fanburg BL. The 5-HT transporter transactivates the PDGFbeta receptor in pulmonary artery smooth muscle cells. Faseb J. 2007; 21: 2725-2734.
- Kashishian A, Kazlauskas A, Cooper JA. Phosphorylation sites in the PDGF receptor with different specificities for binding GAP and PI3 kinase in vivo. EMBO J. 1992; 11: 1373-1382.