
Case Report
Austin J Clin Ophthalmol. 2024; 11(1): 1174.
Case Report of a Child with Harlequin Syndrome and Nance-Horan Syndrome
Breen HA¹; McLoone E¹; Jackson AJ²*
¹Department of Ophthalmology, Royal Victoria Hospital, Belfast, UK
²Northern Ireland Clinical Research Network, Belfast Health and Social Care Trust, UK
*Corresponding author: Jackson AJ Ophthalmology Department, Royal Victoria Hospital, 274 Grosvenor Road, Belfast, BT12 6BA, United Kingdom. Tel: 03005550114 Email: Jonathan.jackson@belfasttrust.hscni.net
Received: December 06, 2023 Accepted: January 19, 2024 Published: January 26, 2024
Abstract
We present the case of a child with idiopathic Harlequin Syndrome and X linked recessive Nance Horan Syndrome, the first case that we are aware of with both rare syndromes occurring simultaneously.
Nance Horan Syndrome causes congenital cataract and variable intellectual disability with facial and dental anomalies. Harlequin Syndrome is characterized by hemifacial flushing due to contralateral loss of sympathetic innervation to the skin.
The co-presentation of these two conditions posed a diagnostic challenge due to iatrogenic post-lensectomy pupillary changes.
Keywords: Nance Horan Syndrome; Harlequin Syndrome; Facial flushing; Congenital cataract; Iatrogenic miosis; c.4449C>G p.(Tyr1483Ter)
Abbreviations: NHS: Nance Horan Syndrome; T2-T3: thoracic vertebral level 2-3; CCFDN: Congenital cataracts, Facial Dysmorphism and Neuropathy; HCC: Hypomyelination and Congenital Cataracts
Introduction
Harlequin Syndrome is a rare condition caused by unilateral autonomic dysregulation affecting the face and sometimes the arms and trunk [1,2]. Patients demonstrate episodic, often striking, hemifacial flushing and diaphoresis with an abrupt demarcation at the midline. It is usually precipitated by exercise, heat or emotion but has also been observed during the sleep/wake cycle in infancy [3]. The skin on the affected side is unable to flush or perspire due to lack of autonomic vasomotor and sudomotor innervation [1-3]. The contralateral skin experiences compensatory sympathetic hyperstimulation resulting in excessive flushing and perspiration [1].
Harlequin Syndrome can be idiopathic or secondary [2,4]. Secondary Harlequin Syndrome can be caused by lesions of the sympathetic pathway that run from the hypothalamus to the lateral horn of the spinal cord, travel via T2-T3 nerve roots to the sympathetic chain and extend onto the internal and external carotid arteries via the superior cervical ganglion [1-3]. Thus traumatic, compressive, inflammatory, ischemic or iatrogenic insults at any part of this sympathetic pathway can result in Harlequin syndrome with or without other dysautonomic syndromes such as Horner’s, Holmes-Adie and Ross syndromes. [2-7].
Nance-Horan Syndrome (NHS) is a rare X-linked recessive disorder characterized by congenital cataract, dysmorphic facies and dental anomalies [8-10]. Microcornea, microphthalmia, strabismus, nystagmus and moderate intellectual disability are also associated [8, 9]. As it is X-linked, it tends to present with a more severe phenotype in affected males. Female heterozygotes are variably affected and can be asymptomatic. They display a classic lens opacity of variable severity centered on the posterior Y-suture [11].
Case Report
We present the case of a young girl with both Harlequin Syndrome and symptomatic Nance-Horan Syndrome. The authors are not aware of another documented case of these two syndromes simultaneously affecting one patient.
Our patient presented to the Royal Victoria Hospital Pediatric Ophthalmology Department in Belfast, United Kingdom, as an infant in 2018 for screening due to a maternal family history of congenital cataract.
She had a visually significant lens opacity in her left eye and a mild cataract in her right eye. She does not exhibit any other features of NHS.
She underwent left lensectomy at two months of age with subsequent aphakic contact lens correction. She required occlusion therapy following left lensectomy to stimulate left visual development. At six months old she was noted to have a small left esotropia and manifest nystagmus. At two and a half she developed a small right esotropia that was felt to be related to deterioration of right vision due to progression of her right cataract. She required right lensectomy with aphakic spectacle correction at the age of two years and ten months.
After her primary lensectomy in 2018 genetic testing identified a mutation in the NHS gene described as c.4449C>G p.(Tyr1483Ter). At that time the mutation was not known to be pathogenic. This mutation has since been identified in another family with NHS and upgraded to “likely pathogenic”. NHS was therefore felt to be the underlying unifying cause of her and her mother’s congenital cataracts.
At eighteen months old, fourteen months after her left lensectomy and sixteen months prior to her right lensectomy, her mother observed two episodes of hemifacial flushing involving her left brow, cheek and upper lip. These lasted for a few hours and spontaneously resolved. The patient was not demonstrably upset or overheated but we can speculate that she may have been undertaking exercise as part of her usual daily activities at the time of onset. The patient was otherwise well with no systemic or neurological symptoms (Figure 1).
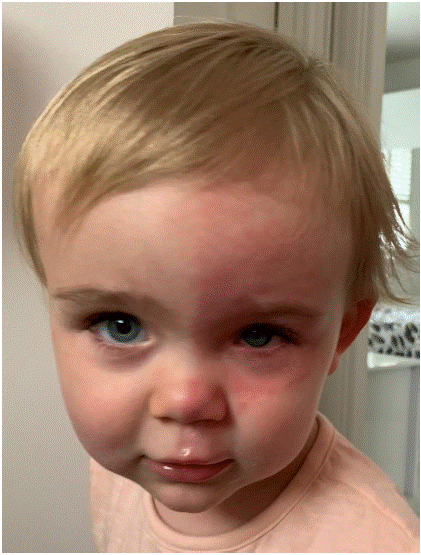
Figure 1: Left unilateral facial flushing.
The striking hemifacial flushing is highly suggestive of Harlequin Syndrome and therefore our patient underwent ophthalmological assessment seeking any additional signs of sympathetic dysregulation. Pupillary examination demonstrated a left miosis which was similar in scotopic versus photopic conditions and not associated with ptosis (Figure 2).

Figure 2: Left Miosis.
Up to 50% of cases of Harlequin Syndrome have an associated Horner’s syndrome [3,13]. In these cases, there is miosis, ptosis and anhidrosis on the contralateral side to the facial flushing because unilateral autonomic interruption causes miosis, ptosis, anhidrosis and vasoconstriction. This is not the case with our patient who demonstrated left miosis and left hemifacial flushing.
She underwent magnetic resonance imaging of her head, neck and lung apices which did not demonstrate any structural abnormality of the sympathetic pathway. Thus, her left miosis was felt to be unrelated to her Harlequin Syndrome and is likely a consequence of her previous left lensectomy.
Her birth history was of vacuum assisted delivery following induction of labor. It is possible that her instrumental delivery resulted in nerve injury that caused her isolated Harlequin Syndrome, however this is felt to be unlikely. She had no history of head trauma or of acceleration-deceleration injuries to her neck. In the context of unremarkable neuroimaging and the absence of any significant medical, surgical or trauma history her Harlequin Syndrome was deemed idiopathic.
Discussion
Harlequin Syndrome and Nance Horan syndrome are both so rare that there is no estimated incidence of either in the literature [4,8]. Following a literature search of Pubmed and Google Scholar we believe this to be the first published account of the two apparently unrelated syndromes occurring in one patient.Harlequin Syndrome is characterized by hemifacial flushing and sweating due to sympathetic hyperstimulation of dermal blood vessels and sweat glands. This arises due to contralateral denervation of these skin appendages either as a result of an insult to the sympathetic nervous system in the affected area or of idiopathic loss of sympathetic innervation.
Nance Horan Syndrome is an X linked recessive condition caused by mutations affecting the NHS gene on the p arm of the X chromosome. Approximately 60 NHS families have been identified worldwide and about 40 causative mutations affecting Xp22.13 have been recorded in the Human Gene Mutation Database [14]. The mutation affecting this patient, c.4449C>G p.(Tyr1483Ter), has been identified in one other family to date. NHS is a phenotypically highly variable syndrome which, particularly in carriers, can have subclinical manifestations and can be uncovered incidentally when investigating relatives of a proband. Features include congenital cataract, dysmorphic facies, dental anomalies, microcornea, microphthalmia, strabismus, nystagmus and moderate intellectual disability [8,9,12,14].
Following a Medline literature review and discussion with the Clinical Genetics Department in the Belfast Health and Social Care Trust the authors are not aware of any genetic link between the NHS gene and autonomic or somatic nervous system dysfunction. It is worth considering that there are a few genetic syndromes such as the autosomal recessive CCFDN (Congenital cataracts, Facial Dysmorphism and Neuropathy) and the autosomal recessive HCC (Hypomyelination and Congenital Cataracts) that demonstrate both nervous system disorders and congenital cataracts [15,16]. Thus, it is not impossible that a genetic foundation for our patient’s sympathetic nervous system dysfunction might be demonstrated as advances are made in clinical genomics. However, currently, there is no unifying genetic explanation for both Nance Horan Syndrome and Harlequin Syndrome co-existing as in this case.
This patient has no known family history of neurodevelopmental disorders or primary autonomic dysfunction. She has no other past medical history. Within the limits of our current understanding, we propose that her two syndromes are unrelated. Her NHS phenotype is mild because she is heterozygous for the NHS mutation and female carriers of X linked recessive conditions only express a proportion of the mutated gene. This is because, by lyonization, only one X chromosome is expressed in each of her somatic cells and therefore she has approximately a 50% chance of expressing a normal Xp22.13 gene in each somatic cell. As a germline heterozygote (inherited from her mother rather than occurring sporadically) with a pathogenic mutation her daughters have a 50% chance of becoming heterozygotes like her and her sons have a 50% chance of becoming affected individuals. This is important because affected males tend to have a more severe phenotype with dense congenital cataracts and intellectual disability [8,9,11].
Conclusion
This is the only published case known to the authors of a patient with both Idiopathic Harlequin Syndrome and Nance Horan Syndrome. The two extremely rare syndromes are not felt to be related. Her left miosis posed an interesting diagnostic challenge as miosis can result from sympathetic dysregulation: however, the laterality did not support a neurogenic miosis and thus it has been attributed to iatrogenic pupillary changes following lensectomy surgery.
References
- Elboukhari K, Baybay H, Elloudi S, Douhi Z, Mernissi FZ. Idiopathic harlequin syndrome: a case report and literature review. Pan Afr Med J. 2019; 33: 141.
- Tascilar N, Tekin NS, Erdem Z, Alpay A, Emre U. Unnoticed dysautonomic syndrome of the face: Harlequin syndrome. Auton Neurosci. 2007; 137: 1-9.
- Kang J, Zafar M, Werner K. Child Neurology: an infant with episodic facial flushing. A rare case and review of congenital harlequin syndrome. Neurology. 2008; 91: 278-81.
- Genetic and Rare Diseases Information Centre. Harlequin syndrome. 2021.
- Biondi A, Persiani R, Zoccali M, Rausei S, Cananzi F, D’Ugo D. Harlequin syndrome. Ann Thorac Surg. 2009; 88: 304.
- Wasner G, Maag R, Ludwig J, Binder A, Schattschneider J, Stingele R et al. Harlequin syndrome – one face of many etiologies. Nat Clin Pract Neurol. 2005; 1: 54-9.
- Kim JY, Lee MS, Kim SY, Kim HJ, Lee SJ, You CW, et al. A pediatric case of idiopathic Harlequin syndrome. Korean J Pediatr. 2016; 59: S125-8.
- Toutain A. National organisation of rare disorders; 2017. Nance Horan Syndrome.
- Genetic and Rare Diseases Information Centre. Nance Horan syndrome. 2015.
- Toutain A. Orphanet rare diseases. 2007. Nance Horan Syndrome. 2021.
- Yeong J. Gene vision; 2020. NHS Gene Overview. 2021.
- Sharma S, Datta P, Sabharwal JR, Datta S. Nance-Horan syndrome: A rare case report. Contemp Clin Dent. 2017; 8: 469-72.
- Bremner F, Smith S. Pupillographic findings in 39 consecutive cases of harlequin syndrome. J Neuroophthalmol. 2008; 28: 171-7.
- Li H, Yang L, Sun Z, Yuan Z, Wu S, Sui R. A novel small deletion in the NHS gene associated with Nance-Horan syndrome. Sci Rep. 2018; 8: 2398.
- Rossi A, Biancheri R, Zara F, Bruno C, Uziel G, van der Knaap MS, et al. Hypomyelination and Congenital Cataract: neuroimaging features of a novel inherited white matter disorder. AJNR Am J Neuroradiol. 2008; 29: 301-5.
- Tournev I, et al. Congenital Cataracts facial dysmorphism neuropathy (CCFDN) syndrome – clinical, neuropathological and genetic investigation. Acta Myol. 2001; 20: 210-9.
- Hainline BC, Sprunger DC, Plager DA, Neely DE, Guess MG. Reverse amblyopia with atropine treatment. Binocul Vis Strabismus Q. 2009; 24: 25-31.
- Patil PA, Meenakshi S, Surendran TS. Refractory reverse amblyopia with atropine penalization. Oman J Ophthalmol. 2010; 3: 148-9.