
Case Report
Austin J Clin Ophthalmol. 2021; 8(1): 1113.
Congenital Anophtalmia: Two Cases Report
Hassan M*, Ahmed B, Chraibi F, Abdellaoui M and Benatiya D
Department of Ophthalmology, Faculty of Medicine and Pharmacy of Fez, Morocco
*Corresponding author: Moutei Hassan, Department of Ophthalmology, Faculty of Medicine and Pharmacy of Fez, Morocco
Received: January 21, 2021; Accepted: February 09, 2021; Published: February 16, 2021
Abstract
Anophthalmia is a rare condition that is estimated to have an incidence of 1 in 10 000 live births. These ocular malformations may be unilateral or bilateral, and may be isolated or occur with other malformations as part of a syndrome. The etiology is heterogeneous and remains unknown. Prenatal diagnosis, which is now available, allows for the early diagnosis of these diseases and the identification of other malformations or signs that may prejudge the prognosis, which must be carefully explained to parents. The prognosis is variable and depends on severity, associated abnormalities and the underlying genetic cause. Reported cases of anophthalmia generally represent severe microphthalmia; true primary anophthalmia is rarely compatible with life secondary to associated brain abnormalities.
Introduction
Congenital anophthalmia is defined by the absence of formation or regression of the primary optic vesicle. It is one of the main causes of congenital blindness and represents 3 to 11% of blindness in children [1]. Anophthalmia is a rare congenital condition, less than one case in 10,000 births. It is most often unilateral and usually occurs as a result of a genetic syndrome, maternal infection, teratogenic exposure or vitamin deficiency. Most cases are sporadic, but an autosomal recessive form has been described.
Diagnosis is clinical, obtained by ophthalmological examination showing the complete absence of the globe or decreased ocular size relative to the contralateral eye. It can be isolated, but is frequently associated with other oculo-orbital, cranioencephalic or even general malformations. Consequently, in all cases of congenital anophthalmia, it is important to conduct an exhaustive clinical, biological and imaging assessment in order to diagnose the associated lesions, but above all to establish the diagnosis, to plan adequate management and to determine prognostic parameters. Differential diagnoses include cryptophthalmos (or ablephary), cyclopia (also cyclocephaly or synophthalmia) congenital cystic eye and extreme cases of microphthalmia.
It must be treated early; it is heavy and often deceiving. They represent about 1.7 to 1.8% of the cases of reconstructive surgery in plastic surgery and ocular prosthesis laboratories [2]. It requires the use of conformators with an increasing size, orbital expansion techniques using pneumatic prostheses or bone surgery.
Methods
We report two observations referred to the Department of Ophthalmology at the University Hospital Center of Fez for treatment between 2017-2019. The study was conducted in accordance with the principles of the Declaration of Helsinki.
Results
Case 1: An 8-week-old infant, seen in consultation for missing eyeballs. The mother was primiparous at 37 years of age, the pregnancies were normal; their births were term births (38 weeks), spontaneous and eutocic. The mother had no reported taking medication, alcohol, tobacco, or drugs during pregnancy.
The initial ophthalmic exam revealed small palpebral fissures with absence of both eyes and other eye structures. Besides the congenital anophthalmia finding, the patients had no other physical manifestations of malformations, and systemic pathologies were discarded at birth (Figure 1). As a history, prenatal laboratory tests revealed no primary toxoplasmic or rubella infections during pregnancy. The mother was previously immunized. No prenatal ultrasound was performed.
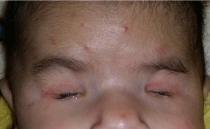
Figure 1: Photographic image showing bilateral anophthalmia.
An ocular ultrasound, a general check-up, a Cerebral Tomodensitometry (CT) and a karyotype were prescribed:
• The ocular ultrasound shows the bilateral existence of rudiments of transonorous eyeballs without crystalline lenses.
• The diagnosis was confirmed with an ocular computer tomography scan that showed under development of both globes within the orbit with a conclusion of bilateral anophthalmia; a brain CT was normal (Figure 2).
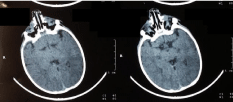
Figure 2: Scanographic images objectifying a bilateral anophthalmia.
• Antibodies for Toxoplasma, Rubella and Cytomegalovirus were negative.
As the case of bilateral anophthalmia was confirmed. Child was sent to department of Trauma and Maxillofacial Prosthetics for a further evaluation and possible surgery. Surgery was performed and further prosthetic treatment was done.
Case 2: A 1-year-old infant, seen in consultation for missing eyeball. This was a second pregnancy from healthy parents. Mother reported that she has lost her first pregnancy, but didn’t report the reason.
The mother had antenatal care in local health center. The pregnancies were normal; their births were term births (39 weeks), spontaneous and eutocic. There was no history of congenital infections like toxoplasma or rubella or any significant teratogenic drug intake during pregnancy. No history of fever, body rashes and no exposure to X-rays. She denied any other medication, illicit drug, alcohol, or tobacco exposure. There was no family history of anophthalmia/ microphthalmia or significant neurological anomalies.
During ophthalmological examination, we found deeply set orbit, small palpebral fissures and livid hemorrhagic cyst s on both lower lids. The eyeball could not be felt on palpation. There was no associated dysmorphic facies or other neurological abnormalities (Figure 3).
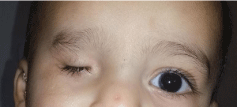
Figure 3: Photographic image showing unilateral anophthalmia.
An ocular ultrasound, a general check-up, a cerebral tomodensitometry and a karyotype were prescribed:
• The ocular ultrasound shows the unilateral existence of rudiments of transonorous eyeballs without crystalline lenses.
• The CT scans objectifying a unilateral anophthalmia; a brain CT was normal
• Antibodies for Toxoplasma, Rubella and Cytomegalovirus were negative (Figure 4).
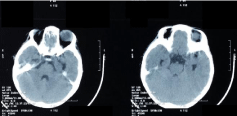
Figure 4: Scanographic images objectifying a unilateral anophthalmia.
• The karyotype was not performed for the both clinical cases.
He was sent from the Department of Ophthalmology to the Department of Traumatology and Maxillofacial Prosthesis to begin rehabilitative treatment.
Discussion
Anophthalmia is a rare malformation which incidence varies according to the authors: it is about 21.34 per 100,000 births according to a Spanish survey [3] and 10 per 100,000 births according to a British study by Busby et al., [4] These abnormalities can be unilateral or bilateral. The frequency of anophthalmias is not influenced by race or gender. Bilateral forms are extremely rare, even exceptional.
Congenital anophthalmia can be analyzed and classified in two ways:
• From an anatomical point of view, one must distinguish between “true” anophthalmia and “true” desembryology, which reflects a cessation of development or degeneration of the primary optic vesicle. Only a histological examination can confirm this.
• From a clinical point of view, clinical anophthalmia (or extreme microphthalmia) must be separated from extreme microphthalmia resulting in a cessation of development at the secondary optic vesicle stage. With a very poor prognosis, from other more moderate microphthalmia with a less severe prognosis.
From this clinical case, some embryological reminders appear necessary in order to better understand the relationships between malformations of the eye, those of other structures of the face, cervical and cerebral malformations.
Anophthalmia results from the cessation of the development of the globe during organogenesis. The development of the face begins in the fourth week of gestation and by the fifth week, the eyes appear as an elevation of the lateral face. Congenital anophthalmia begins between the fourth and seventh week of development and is defined as failure of primary optic vesicle growth. Congenital anophthalmia is much more severe when cessation occurs early in the fourth week of gestation and there is no volume replacement in the remainder of the developmental process [3].
Clinical congenital anophthalmia is an extreme microphthalmia resulting from a failure to develop or regression of the primary optic vesicle during embryonic life. Clinically, no ocular structure is seen, but ultrasound, CT scan and nuclear magnetic resonance can show the presence of an outline of the eyeball and the optic nerve [5].
The associated abnormalities vary depending on the fetal syndrome involved, so a detailed anatomical evaluation by ultrasound is crucial. Triploidy and mosaic trisomies 9 and 13 are among the aneuploidies most likely to present with microphthalmia.
The fetal orbits typically are detectable by 11 to12 weeks of gestation. The diagnosis of anophthalmia or microphthalmia is typically a subjective one, although orbital measurements at each gestational age are available.
The coronal view of the fetal face is the best way to evaluate the orbits. This view can show the size and shape of the orbits, their positioning on the fetal face, spacing between the two orbits, and associated facial abnormalities. Most often, diagnosis of these rare but severe ocular anomalies is made because other fetal anomalies are present and the orbital findings are part of a syndrome. Orbital defects are rarely discovered prenatally as isolated findings.
MRIs and CT scans can be used to scan the brain and eye sockets. Radiologists use them to evaluate the internal structures of the globe, the optic nerve and extraocular muscles, and the anatomy of the brain. Both scans show the absence of a globe within the orbit although soft amorphous tissue may be seen.
Diagnostic tests (amniocentesis or chorionic villi sampling) with Microarray Chromosome Analysis (MAC) should be offered in case of anophthalmia. If screening or other ultrasound characteristics suggest common aneuploidy, it is reasonable to initially perform karyotype analysis or fluorescence in situ hybridization with MAC reflex if these test results are normal. Many syndromes are associated with anophthalmia and may be sporadic, autosomal dominant, autosomal recessive or X-linked. If there are additional abnormalities, inbreeding or a family history of a specific condition, gene panel testing or exome sequencing may be useful because MCA does not detect monogenic (Mendelian) disorders. If exome sequencing is continued, appropriate pre- and post-test genetic counseling by a provider experienced in the complexities of genome sequencing is recommended [6]. Maternal infections (rubella), vitamin A deficiency, and teratogenic exposures (thalidomide) have also been associated with anophthalmia; therefore, it is important to obtain a history of maternal exposures and family history. After appropriate counseling, cell-free DNA testing is an option for patients who refuse diagnostic evaluation if routine aneuploidy is suspected.
Etiologies are dominated by chromosomal aberrations. These can occur during chromosomal deletions or independently of any karyotype aberration. Several genes intervening in ocular embryogenesis may be affected by these mutations. Among the monogenic causes, only SOX2 has been identified as a major causal gene [6]. Anophthalmias of toxic origin due to maternal exposure to thalidomide, nitrofen or benomyl during pregnancy, anophthalmias of infectious embryopathies, particularly rubella, and anophthalmias of undetermined etiology are worthy of note [7]. In addition, there are factors that could increase the risk of anophthalmia, such as: Maternal age over 40 [6]. Multiple births, Low birth weight and premature birth. Moreover, this clinical case helps to recall the different stages of congenital anophthalmia. It underlines the interest of classical bidimensional ultrasound performed by trained operators during pregnancy. Bronshtein et al., in their series 82 reported two cases of isolated anophthalmia, both appearing in patients with a family history of anophthalmia syndromes. Interestingly, in both cases, fetal orbits and optic tissues were normal in the first trimester. This encouraged the authors to suggest that in some cases, anophthalmia may occur later in pregnancy and be related to premature occlusion of the hyaloid artery, producing secondary degeneration of the fetal eye. 82 In view of this, it would certainly be prudent to perform scans later in pregnancy if the initial scans are normal in any patient with a family history of microphthalmia or anophthalmia.
The decision of medical termination of pregnancy for suspected fetal anophthalmia during ultrasound monitoring of pregnancy is extremely delicate. It requires a complete additional assessment in order to specify the stage and prognosis of this malformation, taking into account the functional and aesthetic prejudices that result. The aim of this check-up will be to determine if it is isolated or associated with a polymalformative syndrome.
The specific finding of anophthalmia or microphthalmia does not generally affect delivery management, although delivery in a tertiary care facility with genetic, craniofacial and ophthalmologic pediatric subspecialty services should be considered appropriate for the clinical outcome.
When choosing a modality to treat an anophthalmic socket, the professional must consider the status of the affected eye, the contralateral eye and the severity of volume deficit, the patient’s age, the patient’s support system and the cause of the loss.
Patients with congenital anophthalmia or severe microphthalmia, without potential vision, must undergo early rehabilitation to “artificially” stimulate orbitofacial growth. Several techniques and materials (endogenous and exogenous) are used to expand orbital volume [8].
However, although there are different types of treatment to replace the lost eye, individual ocular prostheses must be made whenever possible because they have better aesthetic and functional results, provide earlier rehabilitation than complex surgical options, and, in addition, are built considering the characteristics of the patient’s eye socket.
Besides restoring aesthetics and protecting tissues, the aim of globe reconstruction is to rehabilitate patients and to integrate them into society. Facial mutilation and the partial or total loss of vision caused by anophthalmia can be a stigma for both patients and their families. The prognosis is variable and depends on severity, associated abnormalities and the underlying genetic cause. Reported cases of anophthalmia generally represent severe microphthalmia; true primary anophthalmia is rarely compatible with life secondary to associated brain abnormalities.
Conclusion
Congenital anophthalmia and microphthalmia are rare developmental defects of the globe. They often arise in conjunction with other ocular defects such as coloboma and orbital cyst. Diagnosis of anophtlamia is usually clinical, following a complete ophthalmologic examination. A coordinated approach with paediatrics is necessary to manage any associated conditions. Genetic diagnosis and investigations can greatly assist in providing a diagnosis and informed genetic counselling.
References
- Wayback Machine. Disponible sur. 2013.
- Kataev MG, Filatova IA, Verigo EN, Kiriukhina SL. [Potentialities of conservative and surgical treatment of patients with congenital microphthalmia and anophthalmia]. Vestn Oftalmol. 2000; 116: 9-13.
- Romero Caballero MD, Lopez Soler JA, Alcazar Cantos A. [Anophthalmia as an isolated congenital defect]. Arch Soc Esp Oftalmol. 2002; 77: 571-574.
- Compiling a national register of babies born with anophthalmia/microphthalmia in England 1988-94 | ADC Fetal & Neonatal Edition. 2021.
- Pathologie orbito-palpebrale. 2020.
- Verma AS, Fitzpatrick DR. Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007; 2: 47.
- Busby A, Dolk H, Collin R, Jones RB, Winter R. Compiling a national register of babies born with anophthalmia/microphthalmia in England 1988-94. Arch Dis Child Fetal Neonatal Ed. 1998; 79: F168-F173.
- Guindo A, Mariko B, Saye1 G, Sangare A, Abass M, Kamano M, et al. Anophtalmie congénitale bilatérale : a propos d’un cas au CSRef de Kita. jaccr Africa. 2020; 4.