
Case Report
Austin J Clin Ophthalmol. 2022; 9(2): 1128.
A Case of Bilateral Pseudo Papilledema Revealing a Leber’s Hereditary Optic Neuropathy
Aachak M*, Brarou H, Boui H, Jeddou I, Abdellaoui T, El Asri F, Mouzarii Y, Reda K and Oubaaz A
Department of Ophthalmology, Military Hospital, Mohammed V University, Rabat, Morocco
*Corresponding author: Meryem Aachak, Department of Ophthalmology, Military Hospital, Mohammed V University, Rabat, Morocco
Received: April 04, 2022; Accepted: April 27, 2022; Published: May 04, 2022
Introduction
Leber Hereditary Optic Neuropathy (LHON) is a mitochondrial disease caused by mutations in mitochondrial DNA affecting the respiratory complex I and leading to the death of retinal ganglion cells (RGCs) [1]. It is characterized by sudden onset and usually severe bilateral loss of central vision, predominantly in young men [2]. The risk of vision loss is 50% among men and 10% among women who carry LHON primary mutations in the mitochondrial DNA [3]. We report an atypic case of LHON in a young 11 years old girl.
Case Presentation
An 11 years old girl coming from a non consanguineous marriage, with no medical history was brought by her parents to the ophthalmology emergency department for a progressive bilateral decreasing of visual acuity.
Ophthalmic examination revealed a best corrected VA of 1/10 in the right eye and “finger counting” in the left eye.
The slit lamp examination was without abnormalities and the fundus evaluation showed an aspect of bilateral papilledema grade II associated with vessel swelling and tortuosities (Figure 1).
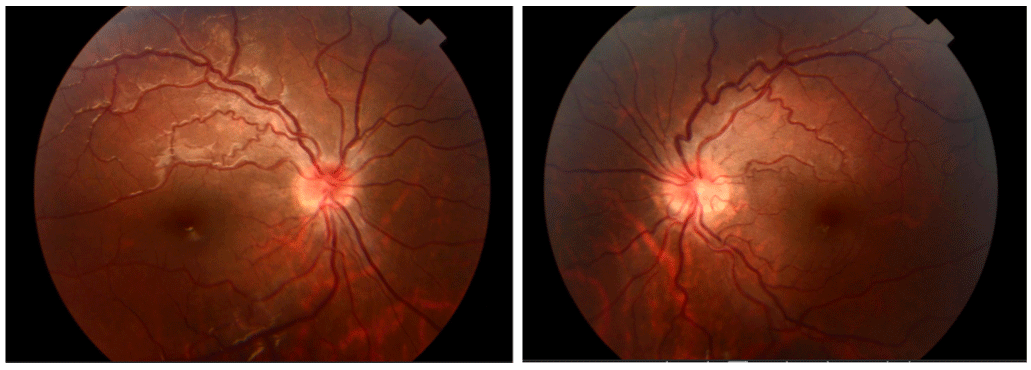
Figure 1: Fundus evaluation showed an aspect of bilateral papilledema grade II associated with vessel swelling and tortuosities.
Retinal fluorescein angiography showed no signs of retention nor papillary fluorescein leakage which refutes the diagnosis of papilledema (Figure 2).
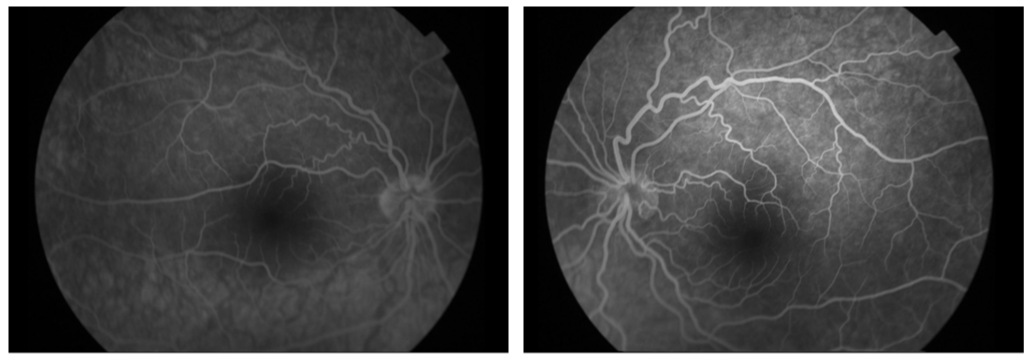
Figure 2: Retinal fluorescein angiography showed no signs of retention nor papillary fluorescein leakage which refutes the diagnosis of papilledema.
Papillary OCT imaging showed a RNFL thickness succeeding 800μm inferior to the optic disc (Figure 3).
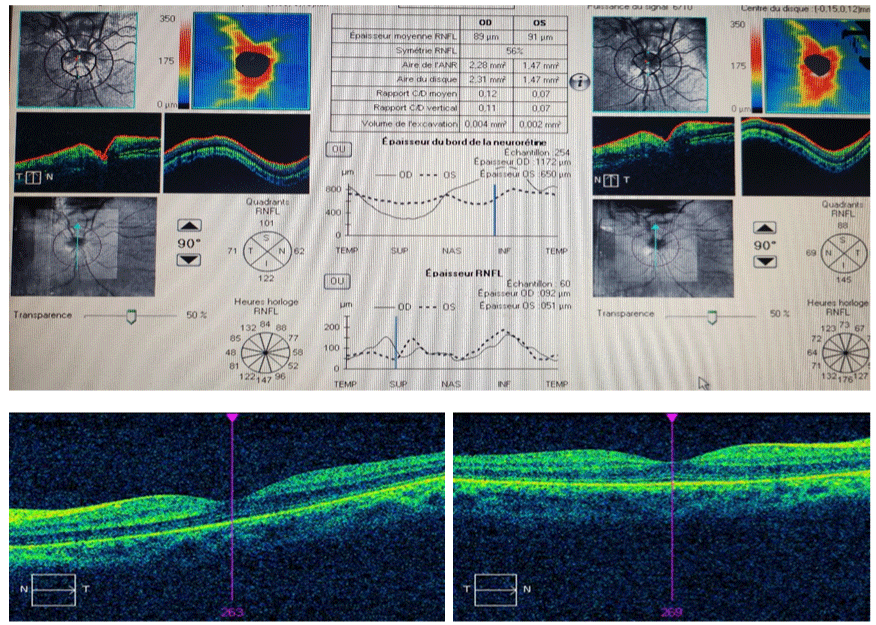
Figure 3: (a) Papillary OCT imaging showed a RNFL thickness succeeding 800μm inferior to the optic disc; (b) normal macular OCT.
An orbito cerebral angio MRI was normal.
VEP in Flash stimulation 30’ and 60’ of arc showed present conduction peaks but hypovolted with impairment of elongated papillary responses (N75) testifying to anterior optic neuropathy (Figure 4).
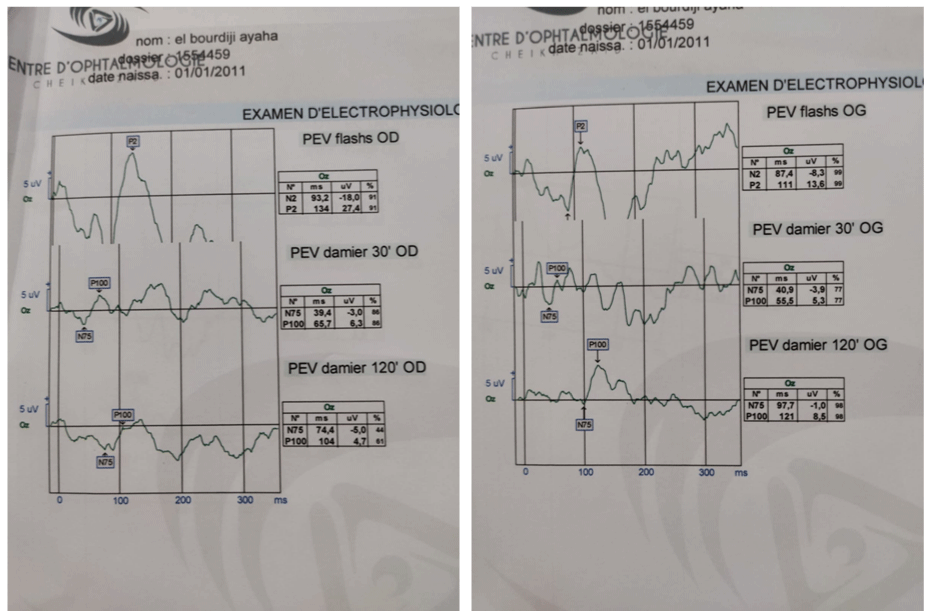
Figure 4: VEP in Flash stimulation 30’ and 60’ of arc showed present conduction peaks but hypovolted with impairment of elongated papillary responses (N75)
testifying to anterior optic neuropathy.
ERG showed normal flicker, photopic and red responses.
Lumbar puncture study was without abnormalities.
Faced with this clinical picture, a Leber optic neuropathy was then suspected and a mitochondrial DNA study was requested objectifying a mutation of G3460A.
Neurological and cardiological consultations were normal. The parents have been informed of the likely evolution of their daughter’s condition.
Discussion
Leber Hereditary Optic Neuropathy (LHON) is a rare disease related to changes in mitochondrial DNA. Its frequency is 1/55000 in northern Europe [3]. Disease onset usually occurs during the second and third decades of life, but the age of onset can start before the age of 14. Barboni found a frequency of infantile forms of Leber’s hereditary optic neuropathy of 11.5% [4].
90% of all LHON cases are due to one of three common mtDNA point mutations, at nucleotide positions G.3460, G.11778, and G.14484, defined as “primary mutations”. Man et al. found the G11778 mutation in 60% of affected families, the G3460A mutation in 33% and the T14448C mutation in 7%. The Alteration of mitochondrial functions plays an essential role in the pathogenesis of hereditary optic neuropathies [5]. In 70% of cases, the onset of the disease is between 11 and 30 years old, with cases described from 2 to 80 years old [2].
LHON typically presents as a painless, subacute, central vision loss in one eye, sequentially spreading to the other eye in weeks or months. Within 1-year, the large majority of affected patients have the second eye involved. Bilateral simultaneous onset occurs in about 25% of patients. During the acute phase, optic disc hyperemia, peripapillary-telangiectatic vessels, vascular tortuosity, and retinal nerve fiber layer (RNFL) pseudo-edema are often detected, even if these features may be subtle, with a slow progression toward blindness. Visual acuity usually reaches a nadir four to six weeks after the first start of symptoms and is severely reduced to 6/60 or less [6].
The characteristic field defect in LHON is a centrocaecal scotoma. Other clinical features include the early impairment of colour perception but, more importantly, pupillary reflexes are preserved and patients usually report no pain on eye movement. Fundoscopy provides other diagnostic clues and in classical cases the following abnormalities can be observed: vascular tortuosity of the central retinal vessels, a circumpapillary telangiectatic microangiopathy, and swelling of the retinal nerve fiber layer. However, it must be stressed that in 20% of LHON cases, the optic disc looks entirely normal in the acute phase [7].
The retinal nerve fibre layer gradually degenerates and after six months optic atrophy is a universal feature of LHON. If a patient is only seen at this stage, it can be difficult to exclude other possible causes of optic atrophy, especially if there is no clear maternal family history [8].
Molecular diagnosis currently shows that about 90% of all LHON cases are due to one of three common mtDNA point mutations, at nucleotide positions m.3460, m.11778, and m.14484 [3]. Considering the clinical signs presented with our patient, the diagnosis of LHON was suspected and then confirmed by the identification of the mitochondrial DNA mutations.
Whilst there is not yet a curative or high-efficacy treatment for LHON, patients with mitochondrial diseases, including LHON, are commonly prescribed ubiquinone (also known as coenzyme Q10), a component of RC3. A molecular analogue of ubiquinone with enhanced bioavailability called idebenone has been recommended. Cree and colleagues reported that significant recovery of vision could be achieved, at least temporarily, with idebenone. Subsequently, in a RHODOS (Rescue of Hereditary Optic Disease Outpatient Study) randomized and placebo-controlled, double-blind trial with 85 patients, the group given high-dose idebenone (900mg) for 24 weeks had better vision than placebo controls without any signs of adverse drug reactions. Patients with early-stage disease appeared to benefit the most. These molecules are thought to counter RC1 deficiency by acting downstream of RC1, effectively bypassing it, to augment ATP production [9].
There is optimism that LHON may be cured, prevented, or at least reduced in severity with gene therapies. The goal of gene therapy for LHON, and other mitochondrial diseases, is to rescue mitochondrial function to an extent that is sufficient to at least relieve the symptoms of, if not cure, the target disease by supplementing intact/wild-type alleles of the dysfunctional gene above the clinical threshold for a normal physiological phenotype [9].
Conclusion
Leber’s optic neuropathy affects men in the majority of cases. It can appear at extreme ages of life, from 1 year to 80 years, and it typically occurs in young subjects. However it must always be kept in mind in case of rapid visual loss unexplained by clinical examination and additional examinations.
References
- J Perruisseau-Carriera, C Debry. Amaurose post-opératoire d’une éthmoïdectomie révélatrice d’une neuropathie optique héréditaire de Leber. Annales françaises d’Oto-rhino-laryngologie et de Pathologie Cervico-faciale. 2019; 136: 330-331.
- M Momtchilova, B Pelosse. Baisse d’acuité visuelle récidivante dans la neuropathie optique de Leber: à propos d’un cas. J Fr. Ophtalmol. 2008; 31: 409-415.
- M Thulliez, B Laudier. Nouvelle mutation de l’ADN mitochondrial dans la neuropathie optique héréditaire de Leber: à propos d’un cas New mitochondrial DNA mutation in Leber’s hereditary optic neuropathy: A case report. Journal Français d’Ophtalmologie. 2018; 41: e293-e299.
- C Orssaud, MP Robert. Neuropathies optiques héréditaires en ophtalmopédiatrie Hereditary optic neuropathies in pediatric ophthalmology. Journal Français d’Ophtalmologie. 2018; 41: 402-406.
- D Milea, C Verny. Neuropathies optiques he´re´ditaires Hereditary optic neuropathies. Revue Neurologique. 2012; 168: 706-709.
- Orssaud C, et al. Raxone dans la neuropathie optique de Leber: retour d’expérience parisienne. Journal Français d’Ophtalmologie. 2019; 42: 269- 275.
- S Leruez. Neuropathies optiques d’origine mitochondriale Les maladies de la mitochondrie, Pratique Neurologique - FMC. 2016; 7: 100-116.
- Meunier A, G Lenaers. Les neuropathies optiques héréditaires: du signe clinique au diagnostic. Journal Français d’Ophtalmologie. 2013; 36: 886-900.
- KD Ayena. Un cas de neuropathie optique héréditaire de Leber chez un mélanoderme africain. Journal Français d’Ophtalmologie. 2017; 40: e287-e290.