
Review Article
Austin J Clin Pathol. 2014;1(1): 1004.
Zhenghong Lin*,Jinping Li
1Department of Pathology, Northwestern University Feinberg School of Medicine, 303 East Chicago Avenue, Chicago, IL 60611, USA. Tel: 312-5033023;
*Corresponding author: Zhenghong Lin, Department of Pathology, Northwestern University Feinberg School of Medicine, 303 East Chicago Avenue, Chicago, IL 60611, USA
Received: February 05, 2014; Accepted: March 10, 2014; Published: March 20, 2014
Abstract
NAD-dependent SIRTuin (SIRT) family deacetylases promote longevity in multiple organisms including yeast, worms, and flies. In mammalian genomes, there are seven members (SIRT1-SIRT7) in the SIRTuin family, with the function of SIRT1 being extensively studied in the past 10 years. Notably, another SIRTuin family member SIRT6, originally identified as mono-ADPribosyltransferase, has recently been drawing more and more attention since it can deacetylate histones and non-histone substrates, and has been emerging as critical regulators in diverse physiological and pathological scenarios including telomere maintenance, chromosome stability, DNA damage repair, glucose metabolism, mammalian aging, life span, and immunity. Dysregulation of SIRT6 leads to metabolic disorder such as type 2 diabetes and cancer. Here we review the recent advances of the function of SIRT6 in immune response, glucose metabolism, and tumorigenesis, and discuss its therapeutic potential in treating cancer.
Keywords: SIRT6; immune response; metabolism; cancer.
Introduction
NAD-dependent SIRTuin (SIRT) family deacetylases, the class III histone deacetylases (HDAC), can extend lifespan of several lower model organisms including yeast, worms, and flies [1]. Mammalian genomes encode seven SIRTuin proteins which share a highly conserved NAD+-binding and catalytic core domain, but have distinct flanking N- and C-terminal extensions [2,(Figure1)]. Except SIRT4, most mammalian SIRTuins had previously been demonstrated to bear a NAD+-dependent protein deacetylases activity. A variety of substrates have been identified for SIRT1 [3]. SIRT4 was originally thought to only have ADP-ribosyltransferase activity [4,5]. However, recently, deacetylation data from David Rauh and colleagues revealed that all seven human SIRTuins have deacetylation substrate candidates including SIRT4 [6].
The subcellular localizations of SIRTuins are quite different [7,(table 1)]. SIRT6 and SIRT7 are nuclear proteins [8,9,(table 1)]. SIRT1, while predominantly in nuclear, can shuttle between cytosol and nuclear in various tissues in response to different stimuli [10]. Whereas, SIRT2 is located mainly in cytoplasm. Different from the above, the other three members, SIRT3, SIRT4, and SIRT5, are primarily found in mitochondria which participate in a variety of metabolic events associated with the mitochondrial activity [11,12].
In the SIRTuin family, SIRT1 was the founding member and had drawn more attention in the past 10 years. Notably, SIRT6 has been increasingly identified as crucial regulators for a variety of physiological and pathological events, ranging from telomere maintenance, chromosome stability, DNA damage repair, mammalian aging, life span, immunity to glucose metabolism and cancer [8,13-20]. Here we highlight the recent progress of SIRT6 studies in immune response, glucose metabolism, and tumorigenesis and discuss the therapeutic potential of SIRT6 modulators in treating cancers.
The ADP-ribosytransferase activity of SIRT6
SIRT6 was reported to have both ADP-ribosytransferase activity [20,21] and deacetylase activity [14,22-24]. Mono-ADP-ribosylation, typically performed by separate families of intra-and extracellular enzymes in vertebrates, is thought to be a general mechanism of reversible protein modification within mammalian organisms [25]. Intracellular mammalian ADP-ribosyltransferases target substrates including molecular chaperone GRP78, translational elongation factor 2, and β-subunit of heterotrimeric G-proteins while extracellular ones generally function in immune system [21,25].
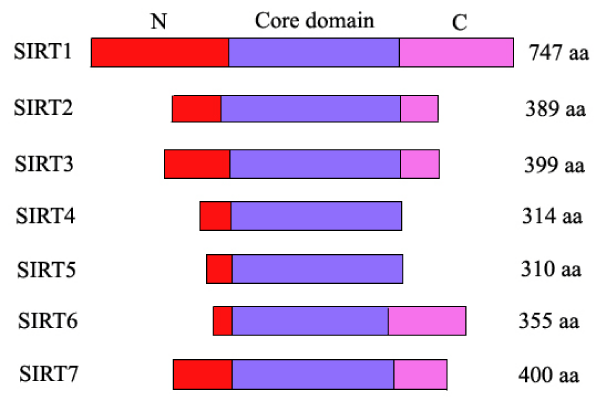
Figure 1: Schematic overview of human SIRTuins. The catalytic core domains (blue boxes) are flanked by distinct N- (red boxes) and C-terminal extensions (pink boxes) in human SIRTuins.
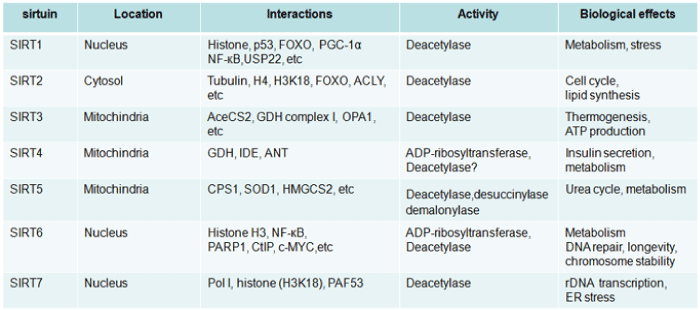
Figure 3: Table 1: Overview of mammalian SIRTuins
Early studies identified SIRT6 as mono-ADP-ribosyltransferase by regulating its own ribosylation [21]. Purified recombinant mSIRT6 catalyzed the robust transfer of radiolabel from [32P]NAD to mSIRT6, suggesting that SIRT6 could regulate its own ADP-ribosylation. Notably, two highly conserved residues within the catalytic core of SIRT6 were required for this reaction. This reaction was likely mono-ADP-ribosylation because only the modified form could be recognized by an antibody specific to mono-ADP-ribose [21].
The auto-regulation of SIRT6 ADP-ribosylation raised the possibility that SIRT6 might target other proteins for ADPribosylation and possibly played an important role in performing its biological activities. In line with its ADP-ribosylation regulation on itself, SIRT6 targeted poly[adenosine diphosphate (ADP)-ribose] polymerase 1 (PARP1) for ribosylation in DNA damage repair [20]. In most cases, ADP-ribosylation of arginine residues in substrates resulted in reversible inactivation of the protein [21,26]. However, mono-ADP-ribosylation of PARP1 by SIRT6 seemed to promote its activity [20]. This might be due to ADP-ribosylation of PARP1 by SIRT6 lying on lysine residue, suggesting a different outcome upon same modification at different amino acid residues. Under oxidative stress, SIRT6 is recruited to the sites of DNA double-strand breaks (DSBs), physically associates with PARP1, mono-ADP-ribosylates PARP1 on lysine residue 521, and stimulates DSB repair [20]. However, whether SIRT6 targets other signaling players for ADPribosylation in certain biological contexts such as immune responses and cancers remains unexplored.
The Deacetylase activity of SIRT6
Although early study reported SIRT6 as a mono-ADPribosyltransferase, hereas a number of other studies implied that SIRT6 functions mainly as a deacetylases to regulate acetylation of histones and non-histone substrates. All seven human SIRTuins have deacetylation substrate candidates including SIRT4 and SIRT6, especially the former which had previously been only demonstrated to have ADP-ribosyltransferase activity [6]. SIRT6 has proved to beable to deacetylate histone 3 at different lysine residues with different outcome. By regulating histone 3 (H3) acetylation, SIRT6 functions as either a life-span modulator, a master regulator of glucose homeostasis, a tumor suppressor, or possibly a regulator of immune responses [14,18,24,27].
SIRT6 was regarded as a life-span modulator by deacetylating histone H3 lysine 9 (H3K9) at NF-?B target gene promoters to attenuate TNFa/NF-?B signaling. Without SIRT6, mammalians can't live long and exhibits aging-like phenotype due to hyperactive NF-?B signaling. Surprisingly, haploinsufficiency of RelA rescues the early lethality and degenerative syndrome of SIRT6-deficient mice, suggesting an implication of SIRT6 regulation in TNFa/NF-?B signaling [24].
In addition, SIRT6 is a guardian for maintaining telomere and chromosome stability. Human SIRT6 protein can modulate telomeric chromatin by deacetylating histone H3 lysine 9 (H3K9) in an NAD+- dependent manner. SIRT6 associates specifically with telomeres and is required for the stable association of WRN, the factor that is mutated in Werner syndrome thus contributing to the propagation of a specialized chromatin state at mammalian telomeres, which in turn is required for proper telomere metabolism and function. Consistent with the above notion, SIRT6 depletion leads to telomere dysfunction with end-to-end chromosomal fusions and premature cellular senescence and exhibit abnormal telomere structures that resemble defects observed in Werner syndrome, a premature ageing disorder. Hence, SIRT6 links chromatin regulation to telomere maintenance and a human premature ageing syndrome [24].
Not only H3K9 but also other lysine(s) at histone 3 including H3K56 can be deacetylated by SIRT6 [22,23]. In S. cerevisiae, acetylation of H3K56 occurs both globally on newly synthesized histones and at specific promoters during S-phase, and regulation of this histone mark is crucial for DNA replication and repair activity such as genomic stability, gene activity and heterochromatin silencing, and histone incorporation into nucleosomal chromatin[28-32]. In mammals, SIRT1 and SIRT2 can deacetylate H3K56Ac, which has recently been linked to stem cell-specific transcriptional networks, chromatin responses, DNA damage, and genomic stability [33-36]. Interestingly, deacetylation of H3K56 by SIRT6 may be cell cycle-dependent, thus revealing a role of SIRT6 in maintaining dynamic changes of H3K56 acetylation levels at telomeric chromatin in the cell cycle progression [23]. Noticeably, although only H3K9 and H3K56 were reported to be deacetylated by SIRT6, however, it cannot rule out the possibility that the acetylation of other lysines on H3 or other histones may be also be targeted by SIRT6 or other SIRTuin members. Consistent with this notion, SIRT7 was reported to be able to deacetylate H3K18ac [37].
Besides histones (H3K9 and H3K56), non-histone substrates including at least DSB resection protein CtIP [C-terminal binding protein (CtBP) interacting protein] and GCN5 can also be deacetylated by SIRT6 [19,38]. Kaidi and colleagues discovered that human SIRT6 plays a central role in promoting DNA end resection, a crucial step in DNA double-strand break (DSB) repair by homologous recombination. Biochemically, SIRT6 interacts with and deacetylates CtIP to promote resection. In line with this notion, SIRT6 depletion impaired the accumulation of replication protein A and single-stranded DNA at DNA damage sites, slowed down rates of homologous recombination, and sensitized cells to DSBinducing agents. Moreover, a nonacetylatable CtIP mutant alleviated the effect of SIRT6 depletion on resection, thus uncovering CtIP as a key substrate by which SIRT6 facilitates DSB processing and homologous recombination and further supporting a role of SIRT6 in promoting genome stability [19]. Interestingly, besides CtIP, the acetyltransferase GCN5 can also be deacetylated by SIRT6. Data from John E. Dominy, Jr. and colleagues suggested that SIRT6 is able to directly bind to GCN5, deacetylate it at K549, as well as induce changes in the phosphorylation of the protein that ultimately yield an increase in GCN5 activity and an increase in PGC-1a acetylation and activity to suppress hepatic gluconeogenesis [38].
Immune response regulation by SIRT6
TNFa/NF-?B signaling plays an important role in the regulation of both the innate and adaptive immune responses and carcinogenesis and the dysregulation of which leads to the onset of tumorigenesis and tumor malignancy [3,39,40]. Deacetylation of H3K9 by SIRT6 at NF-?B target gene promoters raises the possibility that SIRT6 may be involved in normal and/or pathological immune response and tumorigenesis. Consistent with this notion, Van Gool and colleagues discovered that intracellular NAD concentration promotes TNFa synthesis in activated immune cells and SIRT6 regulates TNFa production by acting at a post-transcriptional step in a NAD+- dependent manner [13].
In line with the above data, it was recently reported that SIRT6 promotes TNFa secretion through hydrolysis of long-chain fatty acyl lysine [41]. The crystal structure of SIRT6 reveals that it has a large hydrophobic pocket, which can accommodate long-chain fatty acyl groups. SIRT6 efficiently removes long-chain fatty acyl groups, such as myristoyl, from lysine residues K19 and K20 of TNFa, which modulates TNFa secretion [41]. In this regard, SIRT6 promotion of TNFa secretion seems contrast to previous observation that SIRT6 deacetylates H3K9 at NF-?B target gene promoters, which attenuates TNFa/NF-?B signaling. How to explain this remains obscure.
SIRT6 was also suggested to play an anti-inflammatory role in mice by inhibiting c-Jun-dependent expression of proinflammatory genes [42]. Xiao and colleagues found that SIRT6-null mice developed chronic liver inflammation starting at ~2 months of age, and all animals were affected by 7-8 months of age. Furthermore, deletion of SIRT6 in T cells or myeloid-derived cells was sufficient to induce liver inflammation and fibrosis, suggesting an anti-inflammatory role of SIRT6 in immune responses [42]. Biochemically, SIRT6 interacts with c-Jun and deacetylates histone H3 lysine 9 (H3K9) at the promoter of proinflammatory genes which expression involves the c-Jun signaling pathway. In addition, SIRT6 was also reported to function as a negative regulator of cardiac hypertrophy by interacting with c-Jun and deacetylating H3K9 to suppress the promoter of IGF/ AKT signaling [27].
SIRT6 Regulates Glucose homeostasis and Fat Metabolism
SIRT6 was recently regarded as a master modulator of glucose homeostasis by regulating histone H3K9 acetylation to control the expression of multiple glycolytic genes [18]. Specifically, SIRT6 appears to function as a corepressor of the transcription factor Hif1a to regulate nutrient stress responses. In line with this notion, SIRT6-deficient cells show increased Hif1a activity and exhibit increased glucose uptake with upregulated glycolysis and diminished mitochondrial respiration, thus revealing a role for SIRT6 as a master regulator of glucose homeostasis and may provide the basis for the therapeutic potential of SIRT6 in metabolic diseases, such as diabetes and obesity [18].
It is known to us that under various conditions, mammals have the ability to maintain blood glucose concentration within a narrow range. Dysregulation of hepatic glucose production (HGP) may lead to diabetic hyperglycemia. HGP is dynamically controlled by a signaling/transcriptional network containing PGC-1a, a key mediatorof gluconeogenic enzyme. PGC-1a's activation of gluconeogenic gene expression is determined by its acetylation state, which is reversibly controlled by the acetyltransferase GCN5 and the deacetylase SIRT1. Interestingly, another SIRTuin member, SIRT6, is also involved in HGP by affecting PGC-1a acetylation. Surprisingly, different from SIRT1 and other SIRTuins, SIRT6 positively regulates PGC-1a acetylation by deacetylating and activating the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Consistently, SIRT6 depletion decreases PGC-1a acetylation and promotes HGP and ectopic re-expression suppresses gluconeogenic genes and normalizes glycemia, suggesting a therapeutic potential of SIRT6 in treating insulin-resistant diabetes [38].
Not only glucose homeostasis but was fat metabolism also affected by SIRT6. Liver-specific deletion of SIRT6 in mice causes profound alterations in gene expression, leading to increased glycolysis, triglyceride synthesis, reduced β-oxidation, and fatty liver formation. Clinically, SIRT6 levels in human fatty liver samples were significantly lower than that of normal controls. These data together suggests that SIRT6 plays a crucial role in fat metabolism and has therapeutic potential for treating fatty liver disease, the most common cause of liver dysfunction in humans [43].
The Tumor-Suppressive Effects of SIRT6
In line with its central regulation on telomere maintenance, DNArepair and metabolism, it is not surprising that SIRT6 is involved in cancer metabolism and functions as a tumor suppressor, and subsequently, down-regulation or depletion of SIRT6 protein leads to tumor progression. Except its ability to attenuate TNFa/NF-?B signaling by deacetylating H3K9 at NF-?B target gene promoters [14], SIRT6 also plays an important role in cancer metabolism [16].
Reprogramming of cellular metabolism named Warburg effect during tumorigenesis was known for many years, but the molecular mechanisms regulating this switch remained a mystery. Until recently, Sebastian and colleagues elegantly demonstrated that SIRT6 functions as a tumor suppressor to regulate aerobic glycolysis bymodifying histone acetylation and repressing MYC transcriptional activity in cancer cells [16]. Loss of SIRT6 or transformed SIRT6-deficient cells leads to tumor formation or increased glycolysis and tumor growth, implying a role of SIRT6 in both establishment and maintenance of cancer. Consistently, by using a conditional SIRT6 allele, they showed that SIRT6 deletion in vivo increased the number, size, and aggressiveness of tumors. Moreover, they discovered that SIRT6 was selectively down-regulated in several human cancers. Hence, these observations together highlighted a role of SIRT6 as a critical modulator in cancer metabolism [16].
Our data also showed that protein expression of SIRT6 was reduced in colon cancers, raising the possibility that SIRT6 might play a key role in tumor suppression [17]. Using a proteomic approach, we identified the ubiquitin-specific peptidase USP10, a known tumor suppressor [44], as one of the SIRT6-interacting candidates [17]. Mechanistically, USP10 removes ubiquitin from SIRT6 to protect it from proteasome-mediated degradation. In addition, USP10 enforced the ability of SIRT6 to suppress the transcriptional activity of the c-Myc oncogene, which was recently demonstrated by Sebastian and colleagues [16], thus inhibiting cell-cycle progression, cancer cell growth, and tumor formation [17]. This conclusion was further supported by the observation that a significant reduction in both USP10 and SIRT6 protein expression was monitored in human colon cancers. Hence, previous data and ours together suggest that SIRT6 plays a key role in Warburg effect during the initial stage of tumorigenesis or thereafter maintenance of cancer.
Modulation of SIRT6 Expression and Activity
SIRT6 was, at least, transcriptionally regulated by c-Fos, p53, or a complex containing SIRT1, FOXO3a, and NRF1 [43,45,46,(Figure 2)]. It was recently found that SIRT1 forms a complex with FOXO3a and NRF1 on the SIRT6 promoter and positively regulates expression of SIRT6, which, in turn, negatively regulates glycolysis, triglyceride synthesis, and fat metabolism by deacetylating histone H3 lysine 9 in the promoter of many genes involved in these processes [43].
In addition, SIRT6 was shown to be positively regulated by p53 under standard growth conditions [45]. Interestingly, it seemed that p53 exhibited opposite effects on SIRT1 and SIRT6 levels since compared to wild type mice, p53-/- mice exhibited higher SIRT1 levels [47], but lower SIRT6 levels [45]. It is now known to us that p53 regulates SIRT6 level, however, whether SIRT6 regulates p53 protein level remains a mystery. Further analysis is required to determinewhether p53 level is regulated by SIRT6 by detecting p53 level in SIRT6-/- cells or by other methods.
Moreover, c-Fos was recently reported to be able to induce SIRT6transcription, which repressed survivin by reducing histone H3K9 acetylation and NF-?B activation at the liver cancer initiation stage [46]. Min and colleagues discovered that increasing SIRT6 protein level or targeting the anti-apoptotic activity of survivin at the initiation stage of cancer significantly impaired liver cancer development. Furthermore, a specific expression pattern with increased c-Junsurvivin and attenuated c-Fos-SIRT6 levels was identified in human dysplastic liver nodules, but not in malignant tumours [46]. Thus SIRT6 links histone modification to stress response in liver tumour initiation. This is of great importance since it not only helps us to understand stage-dependent oncogenic mechanisms but also reminds us that it may be targeted to prevent liver tumorigenesis at the cancer initiation stage.
It was previously known that SIRT1 was involved in the regulation of lifespan by nutrient availability [47]. Interestingly, not only SIRT1 but SIRT6 was also found to be involved in lifespan regulation by nutrient condition [45]. Yariv Kanfi and colleagues showed that SIRT6 was regulated by nutrient availability at the post-transcription level. Levels of the mammalian SIRTuin, SIRT6, increased upon nutrient deprivation in cultured cells, in mice after starvation, as well as in rats fed a calorie-restricted diet. The increase in SIRT6 levels was not via an increase in SIRT6 transcription but due to stabilization of SIRT6 protein. These observations implied that at least two SIRTuins, SIRT1 and SIRT6, are involved in the regulation of lifespan by nutrient availability [45].
The protein stability of SIRT6, not only was reported to be regulated by nutrient availability, but also shown to be regulated by the ubiquitin ligase CHIP (carboxyl terminus of Hsp70-interacting protein) or deubiquitinase USP10 [17,48(Figure 2)]. Ronnebaum and colleagues found that CHIP over-expression increases SIRT6 protein expression without affecting SIRT6 mRNA level. In addition, SIRT6 protein half-life is significantly reduced due to an increase in proteasome-mediated degradation in CHIP-deficient cells.Mechanistically, SIRT6 is mono-ubiquitinated by CHIP at K170, which stabilizes SIRT6 and prevents SIRT6 canonical ubiquitination. Furthermore, in CHIP-depleted cells, SIRT6 K170 mutation increases SIRT6 half-life and prevents proteasome-mediated degradation. Most importantly, the absence of CHIP leads to the global decrease in SIRT6 expression and decreased SIRT6 promoter occupancy, which increases histone acetylation and promotes downstream gene transcription. Thus cells lacking CHIP are hypersensitive to DNAdamaging agents, and DNA repair and cell viability can be rescued by over-expression of SIRT6 [48]. Since SIRT6 interacts with both HSP70 [17], and carboxyl terminus of Hsp70-interacting protein, CHIP [48] implies that SIRT6 may form a complex with both proteins and thus links epigenetic regulation to protein quality control to influence pathways that regulate the biology of aging [48]. Notably, further investigations need to be explored to determine which E3 ubiquitin ligase negatively regulates SIRT6 protein stability and determine whether SIRT6 is post-translationally regulated by other enzymes such as MAP3K7/TAK1, which is recently shown to be able to interact with SIRT6 [17].
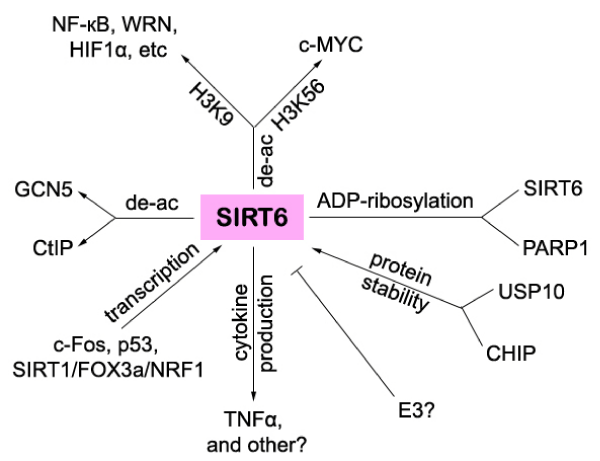
Figure 2: Schematic summary of SIRT6 targets and regulators.
Therapeutic Potential of SIRT6 Modulators
A variety of small molecules have been shown potential for treating human diseases based on their modulation on SIRTuin activity [49,50].Some SIRTuin activators are able to be used to treat diabetes [51] and extend life-span [52], whereas some SIRTuin inhibitors can suppress cancer cell growth and induce apoptosis [53- 61]. However, the small molecular modulators especially activators for SIRT6 are underdeveloped due to its weak enzyme activity and complex biological effects.
Previously, a fluorescence resonance energy transfer (FRET)- based assay where a donor dye and an acceptor dye were connected to an acetyl peptide substrate was developed to screen SIRTuins modulators. However, deacetylation followed by trypsin digest disrupted the FRET signal [62]. Another method was a fluorogenic assay that coupled the deacetylation to the trypsin-catalyzed amide bond hydrolysis to release a fluorescent small molecule, 7-amino-4- methylcoumarin (AMC) [63]. The advantage of the fluorogenic assay using AMC-acetyl peptide is that it can be easily miniaturized and automated for high throughput analysis and has been used to screen deacetylase modulators [51,52].
Recently, by virtue of newly-discovered activity of SIRT5 (demalonylase and desuccinylase) [64] and SIRT6 (defatty-acylase) [41], Hu and colleagues developed a fluorogenic high-throughput assay based on the activity of SIRTuins to screen SIRTuin modulators [61]. They elegantly designed distinct peptides from different SIRTuins for the fluorogenic assay. AMC-acetyl peptides were used for SIRT1, 2 and 3, AMC-succinyl peptides for SIRT5, and AMCmyristoyl peptides for SIRT6 in the fluorogenic assay. The more efficient enzyme activities of SIRT5 and SIRT6 have enabled the development of a high-throughput assay for both proteins since thesenovel activities are several hundred fold higher than the corresponding deacetylase activity [65]. Through this method, they successfully identified a peptide named AcEALPK(MyrK)-AMC for SIRT6, which was thereafter used to screen known SIRTuin inhibitors including nicotinamide [66], SIRTinol [67], AGK-2 [68], Cambinol [53], and Tenovin-1 [69]. Surprisingly, among all the compounds tested, onlynicotinamide showed the best inhibition (57%) at 200 �M, whereas other compounds showed less than 50% inhibition at 200 �M. Most mportantly, it tells us that most known SIRTuin inhibitors cannot inhibit SIRT6 very well and need to be further investigated for SIRT6 inhibition.
Concluding Remarks
The ability of SIRT6 to regulate multiple physiological processes have been recognized and dysregulation of which has been connected to inflammatory disease, metabolic disorder, and even cancers. This raises the possibility that SIRT6 may be targeted for disease therapy. However, how to make use of the pleiotropic effects of SIRT6 (demyristoylation, mono-ADP-ribosylation, deacetylation) for treating disease remains a big challenge to us. Albeit a variety of studies supporting that SIRT6 has tumor suppressive function, however, different voice appeared. Noticeably, in contrast to its down-regulation in certain cancers, SIRT6 was recently reported to be upregulated in lymphoma [70]. Furthermore, the ability of SIRT6 to regulate myristoylation, mono-ADP-ribosylation, and histone or non-histone acetylation, makes it a good target for disease therapy but the pleiotropic effects of SIRT6 must be distinguished before its modulators are applied to certain clinical cases. Finally, although this article is far from satisfaction to cover every aspects of the function of SIRT6, we do hope that it can help our readers to understandthe recent advances of the complex biological effects of SIRT6, thus paving the way for discovering an appropriate approach to treat cancers and other diseases
Acknowledgement
We thank Sinyi Kong for critically reading the manuscript and helpful discussion.
References
- Haigis MC1, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006; 20: 2913-2921.
- Lin Z1, Fang D. The Roles of SIRT1 in Cancer. Genes Cancer. 2013; 4: 97-104.
- Haigis MC1, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006; 126: 941-954.
- Ahuja N1, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, et al. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem. 2007; 282: 33583-33592.
- Rauh D1, Fischer F, Gertz M, Lakshminarasimhan M, Bergbrede T, Aladini F, et al. An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat Commun.2013; 4: 2327.
- Haigis MC1, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010; 5: 253-295.
- Ford E1, Voit R, Liszt G, Magin C, Grummt I, Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006; 20: 1075-1080.
- Tanno M1, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 2007; 282: 6823-6832.
- Li X1, Kazgan N. Mammalian sirtuins and energy metabolism. Int J Biol Sci. 2011; 7: 575-587.
- Verdin E1, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci. 2010; 35: 669-675.
- Van Gool F1, Gall� M, Gueydan C, Kruys V, Prevot PP, Bedalov A, et al. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med. 2009; 15: 206-210.
- Van Gool F1, Gall� M, Gueydan C, Kruys V, Prevot PP, Bedalov A, et al. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med. 2009; 15: 206-210.
- Kawahara TL1, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009; 136: 62-74.
- Kanfi Y1, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012; 483: 218-221.
- Sebasti�n C1, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012; 151: 1185-1199.
- Lin Z1, Yang H1, Tan C1, Li J1, Liu Z1, Quan Q1, et al. USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep. 2013; 5: 1639-1649.
- Zhong L1, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010; 140: 280-293.
- Kaidi A1, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010; 329: 1348-1353.
- Mao Z1, Hine C, Tian X, Van Meter M, Au M, Vaidya A, et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011; 332: 1443-1446.
- Liszt G1, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem. 2005; 280: 21313-21320.
- Yang B, Zwaans BM, Eckersdorff M, Lombard DB. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle. 2009; 8: 2662-2663.
- Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O, et al. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle. 2009; 8: 2664-2666.
- Michishita E1, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008; 452: 492-496.
- Corda D1, Di Girolamo M. Functional aspects of protein mono-ADP-ribosylation. EMBO J. 2003; 22: 1953-1958.
- Riese MJ1, Goehring UM, Ehrmantraut ME, Moss J, Barbieri JT, Aktories K, et al. Auto-ADP-ribosylation of Pseudomonas aeruginosa ExoS. J Biol Chem. 2002; 277: 12082-12088.
- Sundaresan NR1, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med. 2012; 18: 1643-1650.
- Masumoto H1, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005; 436: 294-298.
- Driscoll R1, Hudson A, Jackson SP. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science. 2007; 315: 649-652.
- Han J1, Zhou H, Horazdovsky B, Zhang K, Xu RM, Zhang Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007; 315: 653-655.
- CC1, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, et al. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008; 134: 231-243
- Das C1, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009; 459: 113-117.
- Yuan J1, Pu M, Zhang Z, Lou Z. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle. 2009; 8: 1747-1753.
- Xie W1, Song C, Young NL, Sperling AS, Xu F, Sridharan R, et al. Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol Cell. 2009; 33: 417-427.
- Tjeertes JV1, Miller KM, Jackson SP. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009; 28: 1878-1889.
- Barber MF1, Michishita-Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z, et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature. 2012; 487: 114-118.
- Dominy JE Jr1, Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol Cell. 2012; 48: 900-913.
- Chen LF1, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002; 21: 6539-6548.
- Kiernan R1, Br�s V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, et al. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem. 2003; 278: 2758-2766.
- Jiang H1, Khan S, Wang Y, Charron G, He B, Sebastian C, et al. SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013; 496: 110-113
- Xiao C1, Wang RH, Lahusen TJ, Park O, Bertola A, Maruyama T, et al. Progression of chronic liver inflammation and fibrosis driven by activation of c-JUN signaling in Sirt6 mutant mice. J Biol Chem. 2012; 287: 41903-41913..
- Kim HS1, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010; 12: 224-236.
- Yuan J1, Luo K, Zhang L, Cheville JC, Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell. 2010; 140: 384-396.
- Kanfi Y1, Shalman R, Peshti V, Pilosof SN, Gozlan YM, Pearson KJ, et al. Regulation of SIRT6 protein levels by nutrient availability. FEBS Lett. 2008; 582: 543-548.
- Min L1, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat Cell Biol. 2012; 14: 1203-1211.
- Nemoto S1, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004; 306: 2105-2108.
- Ronnebaum SM1, Wu Y, McDonough H, Patterson C. The ubiquitin ligase CHIP prevents SirT6 degradation through noncanonical ubiquitination. Mol Cell Biol. 2013; 33: 4461-4472.
- Sanchez-Fidalgo S1, Villegas I, Sanchez-Hidalgo M, de la Lastra CA. Sirtuin modulators: mechanisms and potential clinical implications. Curr Med Chem. 2012; 19: 2414-2441
- Villalba JM1, Alca�n FJ. Sirtuin activators and inhibitors. Biofactors. 2012; 38: 349-359.
- Milne JC1, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007; 450: 712-716.
- Howitz KT1, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003; 425: 191-196.
- Heltweg B1, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006; 66: 4368-4377.
- A. R. McCarthy et al., Tenovin-D3, a novel small-molecule inhibitor of sirtuin SirT2, increases p21 (CDKN1A) expression in a p53-independent manner. Molecular cancer therapeutics 12, 352 (Apr, 2013).
- Kalle AM1, Mallika A, Badiger J, Alinakhi, Talukdar P, Sachchidanand. Inhibition of SIRT1 by a small molecule induces apoptosis in breast cancer cells. Biochem Biophys Res Commun. 2010; 401: 13-19.
- Kalle AM1, Mallika A, Badiger J, Alinakhi, Talukdar P, Sachchidanand. Inhibition of SIRT1 by a small molecule induces apoptosis in breast cancer cells. Biochem Biophys Res Commun. 2010; 401: 13-19.
- Lara E1, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, Martinez-Chantar ML, et al. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene. 2009; 28: 781-791.
- Liu G1, Su L, Hao X, Zhong N, Zhong D, Singhal S, et al. Salermide up-regulates death receptor 5 expression through the ATF4-ATF3-CHOP axis and leads to apoptosis in human cancer cells. J Cell Mol Med. 2012; 16: 1618-1628.
- Portmann S1, Fahrner R, Lechleiter A, Keogh A, Overney S, Laemmle A, et al. Antitumor effect of SIRT1 inhibition in human HCC tumor models in vitro and in vivo. Mol Cancer Ther. 2013; 12: 499-508.
- Zhang Y1, Au Q, Zhang M, Barber JR, Ng SC, Zhang B. Identification of a small molecule SIRT2 inhibitor with selective tumor cytotoxicity. Biochem Biophys Res Commun. 2009; 386: 729-733.
- Ota H1, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, et al. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2006; 25: 176-185.
- Hu J1, He B, Bhargava S, Lin H. A fluorogenic assay for screening Sirt6 modulators. Org Biomol Chem. 2013; 11: 5213-5216.
- P. A. Marcotte et al., Fluorescence assay of SIRT protein deacetylases using an acetylated peptide substrate and a secondary trypsin reaction. Analytical biochemistry 332, 90 (Sep 1, 2004).
- Wegener D1, Wirsching F, Riester D, Schwienhorst A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem Biol. 2003; 10: 61-68.
- Du J1, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011; 334: 806-809.
- A. S. Madsen, C. A. Olsen, Substrates for efficient fluorometric screening employing the NAD-dependent sirtuin 5 lysine deacylase (KDAC) enzyme. Journal of medicinal chemistry 55, 5582 (Jun 14, 2012).
- Bitterman KJ1, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002; 277: 45099-45107.
- Grozinger CM1, Chao ED, Blackwell HE, Moazed D, Schreiber SL. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem. 2001; 276: 38837-38843.
- Outeiro TF1, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007; 317: 516-519.
- Lain S1, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008; 13: 454-463.
- Wang JC1, Kafeel MI, Avezbakiyev B, Chen C, Sun Y, Rathnasabapathy C, et al. Histone deacetylase in chronic lymphocytic leukemia. Oncology. 2011; 81: 325-329.