
Review Article
Austin J Clin Pathol. 2014;1(3): 1013.
Intracranial Rosai-dorfman Disease: a Report of Seven Cases with Review of Literature
Qiuping Gui*, Fang Li and Xin Song
Department of Pathology, General PLA Hospital, China
*Corresponding author: Qiuping Gui, Department of Pathology, General PLA Hospital, Central Hospital of China Aerospace Corporation, 28# Fuxing Rd, Haidian District, Beijing 100853, China
Received: June 20, 2014; Accepted: July 09, 2014; Published: July 10, 2014
Abstract
Introduction: Rosai–Dorfman disease (RDD) commonly involves cervical lymph nodes. Although extranodal involvement has been reported in diverse sites, the central nervous system (CNS) manifestation, particularly in the absence of nodal disease is uncommon.
Methods: Specimens from 7 patients with intracranial RDD, diagnosed between 2004 and 2012, were analyzed using clinicopathological and immunohistochemical methods.
Results: These cases analyzed in the present study including six men and one woman, ranging in age from 4 to 60 years, mean age was 35.42 years. These patients presented with headaches, seizures, numbness, or fever. The lesions were multifocal or solitary in the intracranial area. Histologically, the lesions consisted of variable numbers of pale-staining histocytes with emperipolesis. By immunohistochemical analysis, the characteristic histiocytes were positive for S-100 protein and CD68 and negative for CD1a. Treatment consisted of surgical excision, radiotherapy and steroids administration. Follow-up available for 7 cases with intervals ranging from 3 months to 6 years (mean: 15 mo), disclosed one patient recurrence of operative two years after diagnosis and six patients with no evidence of disease progression.
Conclusion: Intracranial RDD is rare, the diagnosis of CNS RDD is challenging. Pathologists should include RDD in the differential diagnosis of dural-based lesions that clinically and radiologically resemble meningiomas and other histiocytosis and granulomatous disease. Therapy remains controversial, but surgical removal of CNS lesions is an effective treatment, and follow-up is necessary to avoid relapses.
Keywords: Rosai-Dorfman disease; Central nervous system; Extranodal sinus histiocytosis with massive lymphadenopathy
Introduction
Rosai-Dorfman disease (RDD), originally called “sinus histiocytosis with massive lymphadenopathy” (SHML), was first described by Rosai and Dorfman in 1969 as a rare benign histiocytic proliferative disorder [1]. RDD generally presents with massive, painless cervical lymphadenopathy, fever, weight loss, and polyclonal hypergamma globulinemia [2]. Histologically, the disease is characterized by lymph node sinus dilatation and large histiocytes demonstrating emperipolesis [3].
Extranodal sites are involved in 30% to 40% of cases, usually affecting the skin, upper respiratory tract, orbit, and testes. Central nervous system (CNS) involvement is unusual. Isolated CNS RDD without other body involvement is even more exceptional, with fewer than 70 reported cases and the vast majority as single case reports. Because of its rarity and morphologic similarities to other benign or malignant disease, intracranial RDD without concurrent lymphadenopathy can be a diagnostic challenge. In this report, we analyzed the clinicopathologic features of 7 cases of extranodal, intracranial RDD. 4 cases of lesions were extraparenchymal and associated with dural involvement, 2 of isolated intraparenchymal lesion, other one both involved dural-based and intraparenchymal regions.
Materials and Methods
7 cases of intracranial RDD diagnosed between 2004 and 2012 were identified in a search of the General PLA Hospital (Beijing, China) surgical pathology files. One patient of 27 years old male occurring in the sellar region has been previously described by the referring pathologist in a case report [4]. Information about clinical manifestations, neuroimaging examinations and follow-up was obtained by reviewing the medical records or by correspondence with physicians.
Formalin-fixed, paraffin-embedded sections were stained with hematoxylin and eosin for routine histological evaluation. Additional 4-micrometer-thick sections were mounted on electrostatic slides and used for immunohistochemical studies. The slides were stained using the EnVision method (Dako, Glostrup, Denmark), an automated immunostainer (Autostainer, Dako), and commercially available monoclonal and polyclonal antibodies. The primary antibodies against the following antigens were used: glial fibrillary acid protein (GFAP; Dako, Glostrup, Denmark; polyclonal, 1 :200 dilution), S-100 protein (Dako, 1 : 2000 dilution), CD1a (Dako, 1 : 50 dilution), MIB-1 (Dako, 1 :200 dilution), CD3 (Dako, 1 : 400 dilution), CD20 (Dako, 1 :400 dilution); and CD68 (Dako, 1 :100 dilution). Special stains for bacteria and fungi including Ziehl-Neelsen strain, periodic acid- Schiff, and Grocott methenamine silver were also preformed in the study.
Results
Clinical features
The clinical data were summarized in Table 1. There were seven men and one woman, ranging in age from 4 to 60 years (mean 35.4 years). The clinical symptoms included headaches (5/7), seizures (3/7), dizziness (1/7), vomiting (1/7) and fever (1/7). None of the patients demonstrated lymph node involvement at presentation.
![]()
Case
Gender
Age
Location
Clinical presentation
Preoperative diagnosis
Treatment
Follow-up
Resection
Radiotherapy
Steroids administration
1
Male
27
Sellar region
Lateral ventricle
Tentorium of cerebellum
Headaches
Loss of vision
Meningioma
Yes
50 Gy in 25 fractions for 30 days
thyroid hormone, corticosteroids
6 years,
Recurrence
2
Male
60
Corpus callosum
Right cerebellum
Headaches, Vomiting
Lymphoma
Yes
No
No
3 months
No recurrence
3
Male
39
Left temporal lobe
Headaches
Seizures
Meningioma
Yes
No
No
2 years
No recurrence
4
Male
54
Left frontal and parietal lobes
Seizures
dizziness
Metastatic carcinoma
Yes
No
No
3months
No recurrence
5
Female
25
Sellar region
Headaches, Fever
Brain abscess
Yes
No
No
Not in detail
No recurrence
6
Male
4
Cerebral hemisphere
cerebellum
Partial seizures
Parasite
Yes
No
No
3 years
No recurrence
7
Male
39
Left occipital lobe
Headaches
Glioma
Y
No
No
2 years
No recurrence
Table 1: Clinical features of patients with intracranial Rosai-Dorfman disease (Compatibility Mode).
Lesions in 4/7 cases were multifocal and located in the supratentorial and infertentorial regions, 3/7cases were isolate separated located in the sellar region, occipital lobe and left temporal lobe of each one. Four cases of lesions were extraparenchymal and associated with dural involvement (Case 1, 3, 5, 7), two of isolated intraparenchymal lesion without dural attachment (Case 2, 4), other one both involved the dura and intraparenchymal regions (Case 6). Clinical features of patients with intracranial RDD are presented in Table 1. On T1-weighted MRI, RDD demonstrated hyperintense or isointense lesions with clear borders with solitary or multiple masses. The lesions had obvious enhancement after gadolinium contrast administration. T2-weighted images showed isointense masses (Figure 1a,b. Figure 2a-c. Figure 3a,b). Before surgery, clinical and radiation diagnoses of patients were meningioma, lymphoma, glioma, metastatic carcinoma, and brain abscess.
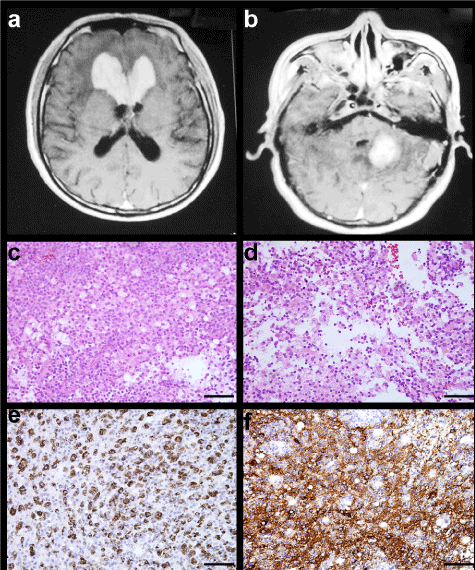
Figure 1: MRI characteristics and light microscopic findings of Case 2. Contrast-enhanced T1-weighted MRI demonstrating multifocal lesions in the bilateralis corpus callosum beak and knee, and right cerebellum (A,B). Sheets of large pale histiocytes with vacuolated eosinophilic cytoplasm admixed with scattered lymphocytes and plasma cells are shown (C,D H&E × 200). The histiocytes are immunoreactive for both CD68 and S-100 protein (E CD68, F S-100 × 200).
All patients underwent surgery with total excision of the mass. Only one of the patients received radiation postoperatively.
Pathological findings
On gross inspection the mass was firm, lobulated, yellow-tan. Microscopic examination revealed a relatively well circumscribed lesion involving the neural parenchyma or dural based. The lesion composed of sheets of large pale histiocytes with vacuolated eosinophilic cytoplasm admixed with scattered lymphocytes, plasma cells and include multinucleate cells (Figure 1c,d Figure 2d, Figure 3c), the lymphoplasmacytic cells were well differentiated without nuclear atypia. Emperipolesis, consisting of well-preserved lymphocytes located within the cytoplasm of histiocytes (Figure 2e, Figure 3c, d), lymphocyte and histiocyte infiltration on along subarachnoid space were noted in the some cases. It was a special feature to showed fibrosis with a polymorphous mixed inflammatory infiltrate composed of histiocytes, mature lymphocytes, plasma cells in the lesion of intracranial RDD compare with lymphoid node. Mitotic figures were infrequent. The adjacent neural parenchyma showed perivascular lymphoplasmacytic cuffing, macrophage infiltration, and reactive gliosis.
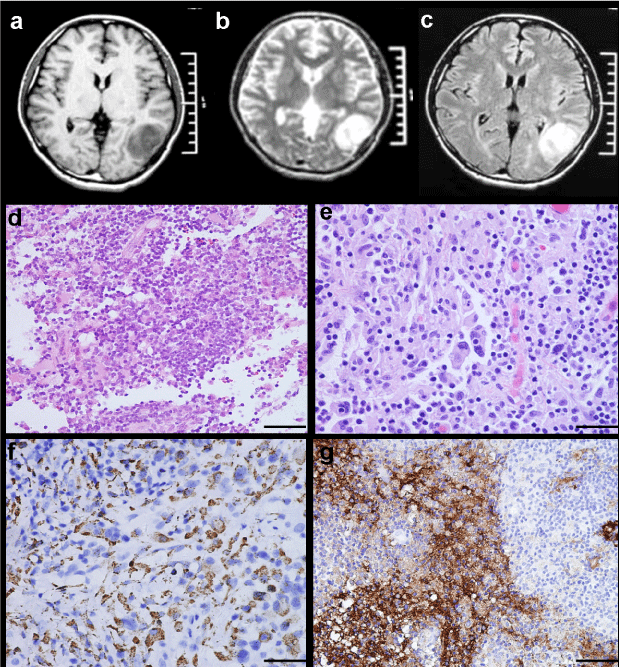
Figure 2: MRI characteristics and light microscopic findings of Case 3. MRI revealing a well-circumscribed lesion in left temporal lobe, which is hypointense to isointense on T1-weighted image (A), isointense to hypointense on T2-weighted image (B), and strongly homogeneous enhanced after contrast administration (C). Scattered large histiocytes intermixed with numerous lymphocytes and plasma cells (D H&E × 200). Multinucleate cells and emperipolesis are evident (E H&E × 400). Strong positivity of histiocytes for CD68 and S-100 protein (F CD68 × 200, G S-100 × 100).
Special stains for bacteria and fungi including Ziehl-Neelsen strain, periodic acid-Schiff, and Grocott methenamine silver were negative. Immunohistochemistry results revealed strong positivity for CD68 and S-100 in the histiocytes, (Figure 1e,f. Figure 2f,g. Figure 3e,f) and lacked immunoreactivity for CD1a in all cases, CD3 and CD20 only focal positive, Occasional cells stained for glial fibrillary acidic protein (GFAP), which were interpreted as entrapped astrocytes within the mass.
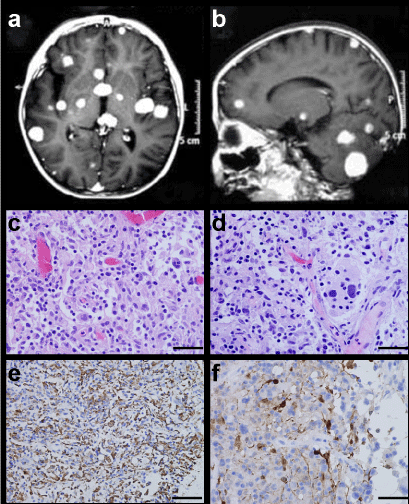
Figure 3: MRI characteristics and light microscopic findings of Case 6. Contrast-enhanced T1-weighted MRI displaying multifocal lesions in the cerebral and cerebellum hemisphere (A,B). Emperipolesis by the histiocytes is clearly seen (C,D H&E × 400). Strong positivity for CD68 and S-100 protein within histiocytes (E CD68 × 200, F S-100 × 400).
Treatment and follow-up
Treatment of all cases consisted of surgical excision. Follow-up, available for 7 cases with intervals ranging from 3 months to 6 years (mean: 15 mo), disclosed one patient recurrence after two years of surgery, other rest of six patients with no evidence of disease progression and without further therapy. The patient of the Case 1 complained of polydipsia and polyuria, associated with weakness and decreased libido after one year postoperatively. MRI did not reveal obvious changes. Laboratory investigation showed evidence of hypogonadia, hypothyroidism, decreased GH, and increased ESR, compatible with hypopituitarism. The patient was treated with prednisone (5 mg/mane, 2.5 mg/nocte) and thyroid hormone suppletion tablet (10 mg once daily) at that time. Three years after surgery, MRI revealed lesions in the sellar region, hypothalamus, superior sagittal sinus, straight sinus, torcular, bilateral introconal orbital, and tentorium of cerebellum, and the patient was readmitted to receive radiotherapy (50 Gy in 25 fractions for 30 days). In addition, the patient was intermittently treated with corticosteroids.
Discussion
Little is known regarding the pathogenesis of RDD. Most studies have suggested that it represents either an autoimmune disease or a reaction to an infectious agent that has yet to be discovered. Molecular studies in two women with RDD have revealed polyclonal X-inactivation patterns, thus implying that this disorder is reactive rather than neoplastic. Some studies have suggested that human herpes virus (HHV-6) and Epstein-Barr virus may play a role in the pathogenesis [5,6].
The central nervous system (CNS) involvement is very rare (less than 5%), and with three fourths in brain and one fourth in spinal cord [7]. CNS lesions tend to present with mass effect, cranial nerve palsies, pituitary dysfunction and seizures [8]. CNS RDD can occur at any age, but most commonly involve patients between 20 and 40 years old with a slight male predominance. Pediatric CNS cases of RDD are extremely rare; the only previously reported 11 cases in pediatric patients [4,9,10]. In our group, a 4-year-old boy complained of partial seizures. Magnetic resonance imaging demonstrated the multiple enhancing lesions in the bilateral cerebrum and cerebellum. At first by considered parasite infection, the final diagnosis of RDD was made by brain biopsy.
Radiologically, the intracranial lesions of RDD tend to be dural based with a predilection for the skull base or orbits the majority of lesions mimic meningioma, because both RDD and meningioma are well-defined, dura-based lesions that do not display local aggression on neuroimaging and exhibit enhancement after contrast administration [6]. Intracranial RDD has been described with lower signal intensity on T2-weighted MR sequences than expected for meningiomas, with variable enhancement [11,12]. Most cases were with a dural-based lesion and extensive systemic involvement showed intraparenchymal invasion, isolate intraparenchymal localization without dural attachment is extremely rare, only 6 cases had reported in the literature [13,14]. Our group of seven cases, four cases were dural-based lesion, two cases were isolate intraparenchyma lesion, other one was involved both dural attachment and intraparenchymal regions. The percentage of lesion with dural attachment and isolate intraparenchyma involvement separate was 71.43% and 28.57%. All of our cases were misdiagnosed as meningioma or others before surgery.
Microscopically, dural-based RDD was similar to intraparenchymal lesion; there are an abundance of histiocytes with abundant pale or foamy cytoplasm and a reactive lymphoplasmacytic cells and eosinophils background. The histopathologic hallmark of RDD is the presence of multiple intact lymphocytes within histiocytic vacuoles, a phenomenon known as lymphophagocytosis or emperipolesis [10,15]. The histiocytes stain positive for S100 and CD68, as well as pan-macrophage antigens. Fibrosis, which was present in the current cases, is normally more pronounced in extranodal sites than in lymph nodes [16], sometimes easy to misdiagnosed with other illnesses.
Because the hallmark feature “emperipolesis” do not present in all cases of RDD, a chronic inflammatory infiltrate may be confused with other entities, such as lymphoplasmacytic meningioma, plasma cell granuloma, langerhans cell histiocytosis, glioma, or a variety of metastatic tumors. Immunohistochemistry helps in confirmation of diagnosis. Immunoreactivity for EMA can make readily differentiates lymphoplasmacytic meningioma from RDD, for EMA is negative in RDD. Plasma cell granuloma is a benign condition that presents as a discrete, dura-based inflammatory mass with associated fibrosis. However, histiocytes are negative for S-100 protein in the plasma cell granuloma, which can differentiate from RDD. Both RDD and Langerhans cell histiocytosis are immunoreactive with S100, but CD1a which constitutes a reliable marker for Langerhans cell histiocytosis, is negative in RDD [17]. Immunohistochemical of GFAP and CK can made readily differentiates between glioma, metastatic carcinoma and RDD. In our study, immunohistochemical results showed CD68- and S100 protein- positive histiocytes with no expression of CD1a, such immunohistochemical criteria aid in the differential diagnosis of RDD.
Rosai-Dorfman disease is considered a benign condition and in most cases surgical resection is the treatment of choice. No specific treatment protocols are available for RDD, and CNS lesions have been noted for their relative persistence. Surgical resection is diagnostic and can relieve symptoms from mass effect. Successful treatment with adjuvant therapy including radiation, chemotherapy or steroids has been reported in cases with CNS disease. However recurrence following removal has been reported. Just like our case 3, he had four times of admitted for radiation treatment to his recurrence lesions.
Petzold et al. found intracranial tumor regrowth or recurrence of symptoms in 14% (4 out of 29) of patients with a mean follow-up of 10.1 years. Of those patients described as “stable”, only 52% had undergone brain imaging at follow-up. They concluded that a 5-year follow-up with brain imaging was essential and advocated local low-dose radiation to treat patients with subtotal resection or recurrence [18,19]. Spontaneous remission is seen in many cases, with occasional stable persistence of disease. A few cases have described a rapidly progressive course leading to death, usually associated with other immunological perturbations. Mortality associated with RDD is reported at 7% [20] Except case 1, all other patients received no further treatment after surgical resection of the mass. Radiation treatment of one recurrence case had been performed and had not got worse after radiation therapy.
In summary, intracranial RDD is rare, the diagnosis of CNS RDD is challenging. The location of intracranial tumors is directly relevant to the preoperative diagnoses. Pathologists should include RDD in the differential diagnosis of dural-based and neural parenchyma lesions that clinically and radiologically resemble meningiomas and other histiocytosis and granulomatous disease and glioma, lymphoma and metastatic carcinoma. Therapy remains controversial, but surgical removal of CNS lesions is an effective treatment, other treatment have including steroid therapy and radiation, and follow-up is necessary to avoid relapses.
Acknowledgment
We are grateful to Dr. Dehong Lu of the Xuanwu Hospital, University of capital medical science for confirming the diagnosis, and also thank for Miss Wei Chen of PLA General Hospital for her valuable technical assistance.
References
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969; 87: 63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990; 7: 19-73.
- Zhang JT, Tian HJ, Lang SY, Wang XQ. Primary intracerebral Rosai-Dorfman disease. J Clin Neurosci. 2010; 17: 1286-1288.
- Wang F, Qiao G, Lou X, Song X, Chen W. Intracranial recurrences of Rosai-Dorfman disease in the sellar region: two illustrative cases. Acta Neurochir (Wien). 2011; 153: 859-867.
- Türe U, Seker A, Bozkurt SU, Uneri C, Sav A, Pamir MN, et al. Giant intracranial Rosai-Dorfman disease. J Clin Neurosci. 2004; 11: 563-566.
- Wang Y, Gao X, Tang W, Jiang C. Rosai-Dorfman disease isolated to the central nervous system: a report of six cases. Neuropathology. 2010; 30: 154-158.
- Bhandari A, Patel PR, Patel MP. Extranodal Rosai-Dorfman disease with multiple spinal lesions: a rare presentation. Surg Neurol. 2006; 65: 308-311.
- Hinduja A, Aguilar LG, Steineke T, Nochlin D, Landolfi JC. Rosai-Dorfman disease manifesting as intracranial and intraorbital lesion. J Neurooncol. 2009; 92: 117-120.
- Di Rocco F, Garnett MR, Puget S, Pueyerredon F, Roujeau T, Jaubert F, et al. Cerebral localization of Rosai-Dorfman disease in a child. Case report. J Neurosurg. 2007; 107: 147-151.
- El Majdoub F, Brunn A, Berthold F, Sturm V, Maarouf M. Stereotactic interstitial radiosurgery for intracranial Rosai-Dorfman disease. A novel therapeutic approach. Strahlenther Onkol. 2009; 185: 109-112.
- Simos M, Dimitrios P, Philip T. A new clinical entity mimicking meningioma diagnosed pathologically as rosai-dorfman disease. Skull Base Surg. 1998; 8: 87-92.
- Zhu H, Qiu LH, Dou YF, Wu JS, Zhong P, Jiang CC. Imaging characteristics of Rosai-Dorfman disease in the central nervous system. Eur J Radiol. 2012; 81: 1265-1272.
- Fukushima T, Yachi K, Ogino A, Ohta T, Watanabe T, Yoshino A, et al. Isolated intracranial Rosai-Dorfman disease without dural attachment--case report. Neurol Med Chir (Tokyo). 2011; 51: 136-140.
- Beros V, Houra K, Rotim K, Zivkovic DJ, Cupic H, Kosec A, et al. Isolated cerebellar intraparenchymal Rosai-Dorfman disease--case report and review of literature. Br J Neurosurg. 2011; 25: 292-296.
- Huang HY, Liang CL, Yang BY, Sung MT, Lin JW, Chen WJ, et al. Isolated Rosai-Dorfman disease presenting as peripheral mononeuropathy and clinically mimicking a neurogenic tumor: case report. Surg Neurol. 2001; 56: 344-347.
- Andriko JA, Morrison A, Colegial CH, Davis BJ, Jones RV. Rosai-Dorfman disease isolated to the central nervous system: a report of 11 cases. Mod Pathol. 2001; 14: 172-178.
- Konishi E, Ibayashi N, Yamamoto S, Scheithauer BW. Isolated intracranial Rosai-Dorfman disease (sinus histiocytosis with massive lymphadenopathy). AJNR Am J Neuroradiol. 2003; 24: 515-518.
- Petzold A, Thom M, Powell M, Plant GT. Relapsing intracranial Rosai-Dorfman disease. J Neurol Neurosurg Psychiatry. 2001; 71: 538-541.
- McPherson CM, Brown J, Kim AW, DeMonte F. Regression of intracranial rosai-dorfman disease following corticosteroid therapy. Case report. J Neurosurg. 2006; 104: 840-844.
- Kattner KA, Stroink AR, Roth TC, Lee JM. Rosai-Dorfman disease mimicking parasagittal meningioma: case presentation and review of literature. Surg Neurol. 2000; 53: 452-457.