
Review Article
Austin J Clin Pathol. 2014;1(4): 1017.
Reactive Oxygen Species and Drug Resistance in Cancer Chemotherapy
Jianli Chen*
Department of Pathology and Laboratory Medicine, Molecular Pathology Laboratory, USA
*Corresponding author: Jianli Chen, Department of Pathology and Laboratory Medicine, Molecular Pathology Laboratory, SIUH, NSLIJ Health System, 476 Seaview Ave, Staten Island 11305, and Feinstein Institute for Medical Research, NSLIJ Health System, Suite 140, 225 Community Drive, Manhasset, NY 11030, USA
Received: August 04, 2014; Accepted: September 03, 2014; Published: September 05, 2014
Abstract
Drug resistance in cancer chemotherapy represents a major obstacle to successful treatment of patients with cancer. Compelling research results suggest that reactive oxygen species (ROS) have been involved in many physiological and pathological processes including apoptosis, tumor hypoxia, regulation of some drug resistance-related proteins, and collateral sensitivity. All of these aspects are associated with drug resistance in cancer chemotherapy. This review will mainly focus on these aspects to sort out the relationship between ROS and drug resistance in cancer chemotherapy.
Keywords: Reactive oxygen species; Drug resistance; Apoptosis; Tumor hypoxia; P-glycoprotein; Dihydrodiol dehydrogenases; Collateral sensitivity; Chemotherapy; Neoplasms
Abbreviation
2-DG: 2-deoxy-D-glucose; AKR: Aldo-keto Reductase; ARE: Antioxidant Response Element; CS: Collateral Sensitivity; DDH: Dihydrodiol Dehydrogenase; DHE: Dihydroethidium; ERK: Extracellular Signal-regulated Kinase; GSH: Glutathione; H2DCFDA: 2',7'-dichlorodihydrofluorescein Diacetate; H2O2: Hydrogen Peroxide; HIF: Hypoxia-inducibble Factor; JAK2: Janus Kinase 2; JNK: c-Jun N-terminal Kinases; MAPK: Mitogen-activated Protein Kinase; MDR: Multiple Drug Resistance; mTOR1: Mammalian Target Of Rapamycin Complex 1; NAC: N-acetylcysteine; NF-κB: Nuclear Factor Kappa B; NF-YA: Nuclear Transcription Factor Y Subunit Alpha; NO: Nitric Oxide; Nrf2: Nuclear Factor Erythroid 2 [NF-E2]-related Factor 2; PAH: Polycyclic Aromatic Hydrocarbon; P-gp: P-glucoprotein; PI3K: Phosphoinositide 3-kinase; PKC: Protein Kinase C; RNS: Reactive Nitrogen Species; ROS: Reactive Oxygen Species; siRNA: Small Interfering RNA; SNP: Sodium Nitroprusside
Introduction
Chemotherapy has become a more and more important weapon of fighting against cancer in clinical practice even although it has been only a little bit more than 50 years since the mid-term of last century, when Gilman et al found nitrogen mustard and applied it to humans after the World War II [1-2]. Currently chemotherapy in single or combined with other methods has been markedly improved survival of patients with cancers, even fortunately able to cure several kinds of tumors like those particularly originated from germ cells, including Choriocarcinoma [3], testes carcinoma [4], and so on. However, drug resistance, no matter intrinsic or acquired resistance, has become the major cause of treatment failure in cancer chemotherapy [5]. To understand and overcome drug resistance, many mechanisms of drug resistance have been proposed [6-8] which can be generally categorized as affecting the pharmacodynamics or pharmacokinetics of anticancer agents [6,8]. Pharmacodynamic resistance includes altered drug target sensitivity, increased DNA repair, and a reduced ability to produce an apoptotic response. Pharmacokinetic resistance usually includes alterations in the stability, metabolism, excretion and distribution of chemotherapeutic drugs at the tumor site [8]. Most recently, the lack of stratification of patients was also been suggested as a cause of resistance to targeted therapy [6].
Based on the observation of an increased GSH-redox cycling capacity in P-glycoprotein (P-gp)-over-expressing multiple drug resistance (MDR) cells, Kramer et al. [9] firstly put forward the theory of "dual mechanism of drug resistance", which brought forth the intriguing suggestion that the MDR should be broadened to include enhanced antioxidant defenses in addition to the typical enhanced drug efflux mediated by P-gp pump. In a sense, this theory actually has implicated the role of ROS in the development and/or the regulation of drug resistance to chemotherapeutic drugs, in particular in solid tumor where probably refers to hypoxia in the central region of tumor tissue, angiogenesis and reperfusion. Since ROS have been demonstrated to be broadly involved in many physiological and pathological processes [10], this review will mainly focus on this point and the relationship between ROS and drug resistance in cancer chemotherapy.
ROS and apoptosis
Apoptosis is an active cell death process initially proposed by Kerr in 1972 [11]. The concept between drug resistance and apoptosis has been well defined, by which drug resistance of chemotherapeutic agents is thought to be the result from inhibition of apoptosis. But this relationship between ROS and apoptosis is not a one-way story [12,13]. The real role of ROS in apoptosis still needs to explore further.
ROS induce apoptosis
In fact, most conventional chemotherapeutic and radiotherapeutic agents kill cancer cells by stimulating ROS generation as at least one part of the mechanisms [13,14]. Even many recently tested compounds with the potential of killing cancer cells were continuously reported to play their role in inducing apoptosis by generating ROS [15-18].
Many different mechanisms on how ROS induce apoptosis have been putting forward. Earlier research results showed that ROS might play a direct killing role in their cytotoxicity of chemotherapeutic agents and recombinant human tumor necrosis factor (rhTNF). Later ROS have been implicated as mediators of apoptosis by activating different caspases or up-regulating death receptor 5 and pre-treatment with N-acetyl-cysteine (NAC) or antioxidative protein can dramatically inhibit apoptosis [19,20]. Recently their molecular mechanisms have been further elucidated. Many different signaling pathways, such as mitogen-activated protein kinase (MAPK) pathway, extracellular signal-regulated kinase(ERK) pathway, phosphoinositide 3-kinase(PI3K) signaling pathway and so on, have been found to get involved in ROS-induced physiological or pathological processes including apoptosis [13,21-23].
ROS level in the resistant cancer cells are usually lower than that in their parental sensitive cells. Our preliminary study by siRNA knockdown and forced over-expressing transfection and other research have found that the resistant cancer cells often show higher level of antioxidants and lower level of ROS than the sensitive parental cell lines [24,25]. So generally speaking from above, ROS are usually considered to increase the chemo-sensitivity by inducing apoptosis [26,27].
ROS inhibit apoptosis
Even though the majority of the body of evidences support ROS induce apoptosis, however, there are also many evidences which showed that ROS could inhibit apoptosis [28,29] and apoptosis could be stimulated by inhibiting ROS with antioxidants [28]. As a matter of fact, this is also easily understandable. Normally, healthy cells are in a biological status characterized by a balance between low steady-state level of ROS and a constant level of reducing equivalents. However, if this balance was broken, usually by an increased ROS level and beyond the cellular ability of returning to the balance, it would cause cellular genomic instability, mutations, and finally even malignancy. Therefore, in this regard, malignancy as a result from apoptosis inhibition, could be caused by ROS stress [13,30,31]. Earlier research finding has already demonstrated this similar point. Caspases (cysteine aspartate proteases) have been well known to be involved in the final stage of apoptotic process; however, ROS could also prevent caspase activation in prolonged or excessive oxidative stress [32]. Recently different ROS species have also been reported to play different roles in ROS-associated apoptosis [13]. Azad et al. reported that superoxide anion played a pro-apoptotic role by causing down-regulation and degradation of Bcl-2 protein through the ubiquitin-proteasomal pathway. In contrast, nitric oxide (NO)- mediated S-nitrosylation of Bcl-2 prevented its ubiquitination and subsequent proteasomal degradation, leading to inhibition of apoptosis [33].
On the other hand, in the most recent years more and more natural plant extracts were found to have the potentials to kill cancer cells. Many these findings support the typical increased ROS-driven apoptosis pathway to kill cancer cells [34,35]. But many others found some extracts decreased ROS level when inducing apoptosis, which implies the reverse role of ROS in apoptotic process. For example, salidroside was found to significantly reduce the proliferation of human lung cancer A549 cells and to induce apoptosis but it decreased ROS generation in A549 cells in a dose- and time-dependent manner [36]. Cho et al reported that butein caused breast cancer cell death by the reduction of ROS production. In their research they also found that NAC, a free radical scavenger, also reduced ROS production and Akt phosphorylation, resulting in apoptotic cell death in butein-sensitive breast cancer cell lines [37]. Furthermore, the inhibition of apoptosis by ROS was reported to be seen in a variety of cell lines from different tissue origins including pancreatic cancer cell, smooth muscle cells, leukemia cells, colorectal cancer cells, and prostate cancer cells [28]. Although the mechanism involved is still controversial, redox status and/or hydrogen peroxide (H2O2) have both been proposed as critical factors [29,38]. This effect of ROS might also be mediated through antiapoptotic redox-sensitive pathways [28,39].
ROS and tumor hypoxia
Hypoxia is a common phenomenon existed in the central region of solid tumors because of insufficient penetration and diffusion of oxygen and nutrition. Before the formation of new abnormal vessels, malignant solid tumors can be only about 1 to 2 mm3 in diameter [40]. Rapidly growing malignant solid tumors are very susceptible to deficiency of nutrients (mainly glucose deprivation) and oxygen (hypoxia) by expanding beyond the perfusion capacity of its blood supply.
ROS and glucose deprivation
Studies showed that during glucose deprivation, decreased peroxide scavenging (by pyruvate and NADPH dependent pathways) could result in increased ROS level (H2O2) by mitochondrial electron transport chain, which has been shown to further cause cytotoxicity, activation of signal transduction (ERK1/2, JNK, and Lyn kinase), and increased expression of genes associated with malignancy (bFGF and c-Myc) in MCF-7/ADR human resistant breast cancer cells [41]. Malignant tumors usually support their growth by stimulating blood vessel development (angiogenesis) [42]. However, blood flow within these new vessels is often chaotic, causing periods of hypoxia followed by reperfusion and consequent generation of ROS. So it is easily understandable about some change of biological characteristics induced by hypoxia and ROS due to serials of genes expression including drug resistance.
Most research in vitro on glucose deprivation employed 2-deoxy- D-glucose (2-DG) to imitate the state of glucose deprivation. 2-DG is a glucose analog molecule which has replaced 2-hydroxyl group by hydrogen. It acts to competitively inhibit the production of glucose- 6-phosphate from glucose. Glucose hexokinase phosphorylates 2-deoxyglucose, trapping the product 2-deoxyglucose-6-phosphate intracellularly and therefore stops further glycolysis. Earlier evidence showed that 2-DG caused cytotoxicity via a mechanism involving perturbations in thiol metabolism and 2-DG was found to cause a 50% of decrease in intracellular total glutathione (GSH) content. Simultaneous treatment with antioxidant NAC protected HeLa cells against the cytotoxicity effects from 2-DG [43]. Currently more and more findings have supported the conclusion that glucose deprivation as well as treatment with 2-DG induces the generation of ROS (including superoxide and H2O2) and subsequently causes cytotoxicity [44-47]. However, in the meanwhile, it was also found that 2-DG could cause chemoresistance by up-regulating the expression of P-glycoprotein through ROS stimulation [48,49]. Actually, our laboratory findings also supported that 2-DG could induced chemoresistance in human ovarian and breast cancer cells by up-regulating the expression of dihydrodiol dehydrogenases (DDHs) and many other different drug resistance-associated target genes(unpublished data) and dihydrodiol dehydrogenases have been demonstrated to regulate the generation of ROS and cisplatin resistance [25].
ROS and oxygen deficiency
For the oxygen deficiency of tumor cells, the most important adaptive mechanism is activation of hypoxia-inducible factor 1 (HIF1). HIF1 consists of two subunits: HIF1α and HIF1β. Under normal oxygen condition, HIF1β is stable but HIF1α is hydroxylated by prolyl hydroxylases. However, when in hypoxic conditions, HIF1α is stabilized and dimerized with HIF1β and regulates the expression of the large number of genes involved in the hypoxic response [50,51]. ROS have been involved in the regulation of HIF1 under various conditions but the role of ROS is still in controversy and the mechanism underlying the HIF-1 regulation by ROS is not completely understood. Insulin and angiotensin II were reported to produce cellular ROS, which were indispensable to increases of HIF- 1a protein translation in normoxia. In human prostate carcinoma cell line, it was found that JNK and JAK2 pathway regulated the ROS-induced HIF1α expression as an upstream of AMPK signaling [52]. Conversely, ROS could also down-regulate expression of P-gp in Nox-1 overexpressing prostate tumor spheroids. Nox-1 is an enzyme to generate intracellular ROS. Pretreatment with free radical scavengers increased the expression of P-gp and HIF1α [53].
As a master switch for hypoxic gene expression, HIF1α could be regulated by H2O2 through several possible molecular mechanisms and it was estimated that about 5% of the human genome comes under HIF-1 control [54]. Multidrug resistance transporter P-gp is one of HIF1α-targeted genes [55-57]. HIF1α -mediated P-gp expression to hypoxia-induced drug resistance has been reported in many different cancer cell lines including gastric cancer, glioma, breast cancer, and colon cancer. Inhibition of HIF1α with specific siRNA was also found to significantly decrease the expression of P-gp and the regulation of P-gp by HIF1α was due to a direct effect of HIF1α binding to P-gp gene promoter [57].
Hypoxia causes ROS generation in solid tumors. Persistent oxidative stress may cause other adaptive responses like up-regulation of antioxidants within tumor cells that confer resistance to apoptosis. All of this up-regulation of anti-ROS defenses may eliminate ROS-induced cytotoxicity and apoptosis to increase the detoxification ability (resistance) of tumor cells to chemotherapeutic agents [58].
ROS and some MDR-associated proteins
ROS and P-gp
P-gp belongs to a family of plasma membrane proteins encoded by the MDR gene(s). Increasing studies show that ROS can regulate expression of P-gp [53,56,57,59]. Both transient and chronic ROS stress were found to up-regulate P-gp expression in rat brain microvessel endothelial cells [48,60] and ROS could induce P-gp expression on RNA, protein and functional levels and this effect could be counteracted by antioxidants. On the other hand, ROS could also work as negative regulator to down-regulate P-gp expression [59,61- 63]. In fact, the recent findings showed that ROS could regulate P-gp expression biphasically. When human colon cancer Caco-2 cells were exposed to low concentration of H2O2 (1μM), P-gp expression was increased; however, when exposed to high concentration of H2O2 (10mM), P-gp expression was decreased [64]. Furthermore, Duan et al. [65] reported that P-gp expression and function could be even biphasically regulated by ROS through a time-dependent mode under the same ROS level stress. Short-term exposure (4- hour) to nitric oxide (NO) donor like sodium nitroprusside (SNP) increased intracellular ROS and reactive nitrogen species (RNS) in human colon cancer Caco-2 cells and impaired P-gp function and expression, whereas long-term exposure (24-hour) stimulated P-gp function and expression. This is compatible with the earlier finding reported by Wartenberg et al. that low levels of ROS down-regulated P-gp expression whereas high concentrations of prooxidants resulted in up-regulation of P-gp in multicellular prostate tumor spheroids [53,56]. Therefore, the final biological effect of P-gp regulation by ROS could be dependent on the duration and the intensity of ROS exposure.
The mechanism that ROS regulate P-gp expression is not completely clear. Recent research showed PI3K inhibitor, p38 inhibitor and PKC inhibitor could reverse the P-gp stimulation induced by long-term exposure to SNP, which suggested that 24- hour exposure to NOx donors stimulated the expression and activity of P-gp via the PI3K/Akt, PKC and p38 MAPK pathways [65]. Wild type p53 is generally known to repress the expression of P-gp through interaction with basal transcription factors, such as TATA-binding protein. Interestingly, mutant p53 is still capable of interacting with TATA-binding protein but is unable to repress MDR1 transcription. On the contrary, mutant p53 has been demonstrated to activate P-gp gene promoter [66]. Furthermore, certain kinases existing in various signaling pathways such as ERK1/2, PKC, SAPK and Akt are activated by ROS exposure. It seems that the NF-kappaB (NF-κB) pathway, in particular, plays a significant part in modulating ROS-mediated P-gp expression. Earlier study showed that up-regulation of P-gp was via NF-κB activation [56]. By depressing NF- κB signaling, H2O2 may still augment P-gp expression when ERK1/2, PKC or SAPK are inhibited [67,68].
ROS and DDHs
DDHs are a superfamily of aldo-keto reductases (AKRs) that normally convert PAH (polycyclic aromatic hydrocarbon) to PAH o-quinones, which is cytotoxic and mutagenic. During the metabolic process of PAH to PAH o-quinones, large amount of ROS and o-semiquinone anion radicals are produced. However, this does not mean that ROS are just only byproduct during this process. Research suggested that DDH1 (AKR1C1) was also reversibly inducible by ROS and multiple classes of xenobiotics. Under the stimulation of oxidative stress with H2O2 and GSH depletion, DDH1 could be induced up to a 10-fold increase. This implies the role of DDH1 as an antioxidant response element in redox systems [25,69].
In view of its inducible antioxidant property of DDHs, our research and other authors have demonstrated that DDHs also contributes to chemotherapeutic resistance in many human cancer cell lines [69-74]. Our recent findings showed that DDHs, particularly DDH1, could directly regulate intracellular ROS level by forced over-expression and siRNA techniques in human ovarian cancer cell lines; DDH1 over-expression attenuated intracellular ROS level and increased resistance to cisplatin; on the other hand, DDHs knockdown by siRNA significantly increased intracellular ROS level and decrease drug resistance to cisplatin [25,75]. Even though cisplatin has been widely known that it can induce intracellular ROS generation, and our findings indicated that nuclear transcription factor Y subunit alpha (NF-YA) can regulate the basal transcription of DDH1 in human ovarian, lung and liver carcinoma cells and the cisplatin-induced CCAAT box-dependent transcription in human ovarian carcinoma cells [76], however, the mechanisms how DDHs are regulated by ROS involved in drug resistance still need to be elucidated further.
A growing body of evidence indicates that ROS are important intracellular signaling regulators in many physiological and pathological processes, including carcinogenesis [13,77] and development of drug resistance to anticancer agents [13,78]. ROS consist of various free radicals, which exert different effects on cellular signaling. By signal transduction, cells communicate with their environment or neighbor cells, connecting extracellular stimuli into specific transcription factors and thereby converting these signals into cellular responses. ROS have been reported to activate the extracellular signal-regulated kinases (ERKs), the c-Jun N-terminal kinases (JNKs), and the p38, which are three subgroups of mitogen-activated protein kinases (MAPKs) (see recent review [79]). Our research group found that treatment of human ovarian cancer cell lines to cisplatin induced increasing intracellular ROS level with H2DCFDA (for H2O2) and DHE (for superoxide) probles and consequently phosphorylated JNK/p38, which was compatible with subsequent levels of apoptosis/necrosis and changes of mitochondrial membrane potential with JC-1 probe; On the other hand, the above changes of cisplatin-induced ROS and subsequent JNK/p38 activation in this pathway could be attenuated by corresponding DDH1 expression level. With external H2O2 and antioxidant NAC further demonstrated the above findings (Figure 1). These findings are also compatible with our previous report about the relationship between ROS and DDHs expression level [25]. DDHs are classic antioxidant response element (ARE) genes that are transcriptionally up-regulated by Nrf2 and Nrf2 was originally identified as protective transcription factor from ROS stress. Chen et al recently reported that knockdown of Nrf2 not only decreased the levels of DDH1, DDH2, and DDH3 mRNA and protein but also reversed oxaliplatin resistance in gastric carcinoma S3 cells [69]. This further suggested the relationship with drug resistance and the inter-regulation between ROS and DDHs.
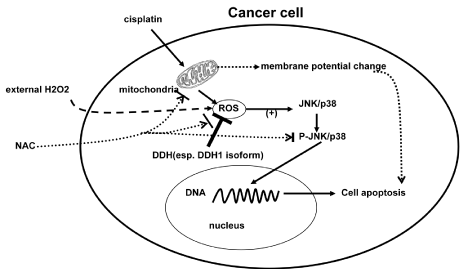
Figure 1: Cisplatin stimulated cancer cell to generate intracellular ROS through mitochondria. ROS subsequently activated JNK/p38 pathway and the phosphorylated JNK/p38 translocated into nucleus and bound to DNA to cause cellular apoptosis, which could also be reflected by the change of mitochondrial membrane potential following cisplatin treatment. Through the same mechanism, external H2O2 exposure could activate the JNK/p38 pathway and antioxidant NAC inhibit it. More interestingly, DDHs, especially DDH1 isotype, as member of intracellular antioxidant system, could directly attenuate intracellular ROS and prevent JNK/p38 pathway from activating and consequent apoptosis to cause chemoresistance.
PI3K-Akt-mTOR1 signaling axis and its regulation by Notch1 was also recently reviewed in resistant T-cell acute lymphoblastic leukemia [80,81]. Lee et al reported that cadmium treatment induced increasing intracellular ROS level and DDH3 expression, which was suppressed by NAC and therefore it was believed that up-regulated DDH3 expression was due to the activation of Nrf2 and the PI3K-Akt pathway by cadmium-induced ROS generation [21]. Furthermore, the activation of mTOR1 pathway by cadmium-induced ROS was also reported by another group [23]. Similarly, Lee et al. also demonstrated that ROS generation and/or activation of PI3K/Akt signaling regulated cell survival and HO-1 expression by Nrf2 transcriptional regulation in sulforaphane-treated human mesothelioma MSTO-211H cells [22]. This is easily understandable since like DDHs, heme oxygenases are also one of the target genes by Nrf2 [82].
Signaling transduction is indeed important since almost all kinds of biological processes cannot go without it. Although more and more molecules in signaling pathways are being as targets for designing new anticancer agents to fight against cancer and chemoresistance, however, these anticancer drugs on new targets could also come to resistance finally [80,83]. Thus unfortunately we might come back to the beginning point after hard hunting. Therefore, creative ideas and innovative thoughts to combat against drug resistance are definitely needed in this field of signal transduction research on chemotherapy.
ROS and collateral sensitivity
Collateral sensitivity (CS) was originally used in 1952 and currently has been extended to broadly describe the observation that the development of resistance to one agent can confer greater sensitivity to another agent than seen in the original parental line [84]. The underlying mechanism of CS has not completely understood. However, the potential role of ROS in CS has been proposed recently [84,85]. Based on the evidences available right now, there are two possible different pathways which might get ROS involving in CS: P-gp-based ATPase stimulation and non-P-gp dependent ROS hypersensitivity (see recent review for details) [84,85].
P-gp-based ATPase stimulation pathway has been supported by earlier studies and recent evidence [85,86]. The non-P-gp dependent pathway is also being gradually demonstrated by more increasing findings. Most recently, Krzyzanowski et al. reported for the first time that BCRP- overexpressing MDCKII-BCRP cells are more vulnerable to ROS. These cells have significantly lower GSH level and decreased activities of glutathione S-transferase and glutathione reductase, implying the possibility of using agents that induce ROS to selectively kill cells overexpressing BCRP [87]. Another group also reported that natural flavonoids and some synthetic derivatives could induce CS activity through GSH efflux (which results in increased intracellular ROS level) in resistant MRP1-overexpressiong cells [88]. As ATP-binding cassette (ABC) efflux transporters, there is substantial overlap in substrate recognition among P-gp, BRCP and MRP1, but BRCP and MRP1 have not been definitively demonstrated to contribute to MDR in patients [84]. Hall et al. recently also demonstrated the drug tiopronin-induced non-P-gp dependent mechanism for CS. They found the CS activity of tiopronin was mediated by the generation of ROS and the inhibition of glutathione peroxidase (GPx) and all these CS activity can be reversed by ROS-scavenging compounds including NAC [89].
Summary
ROS are important mediators in drug resistance of chemotherapeutic agents. In this review, we have discussed recent findings illustrating the correlation of ROS with drug resistance in several aspects. In fact, theoretically drug resistance is an inevitable event as a protective mechanism of cell survival. Therefore, for better fighting against drug resistance, novel points of view or strategies are still needed to dig out the Achilles' heel of chemoresistance to overcome this challenging obstacle in clinical treatment of patients with tumor. The potential role of ROS in collateral sensitivity calls for more attention too.
Furthermore, conventionally we always look at antioxidants as "good guy" since they attenuate ROS produced in normal cellular processes and may protect cells from oxidative damage. However, clinical trials have surprisingly shown that antioxidant supplementation increases the risk of lung and skin cancers. Recent studies on electron transferring reported that, compared to oxidative damage caused by ROS, reductive damage by electron transferring might represent another previously unrecognized important mechanism for DNA damage and could even double the DNA damage caused by oxidative damage from ROS [90,91]. If so, electron transferring and chemotherapeutic agents, reductive damage and chemotherapy and their relationship with chemosensitivity will definitely need to be investigated further for the benefits of patients with cancer in future treatment.
References
- Gilman A, Philips FS. The biological actions and therapeutic applications of the B-chloroethyl amines and sulfides. Science. 1946; 103: 409-415.
- Gilman A. The initial clinical trial of nitrogen mustard. Am J Surg. 1963; 105: 574-578.
- Liu Y, Yang J, Ren T, Zhao J, Feng F, Wan X, et al. The encouraging prognosis of nongestational ovarian choriocarcinoma with lung metastases. J Reprod Med. 2014; 59: 221-226.
- Rijlaarsdam MA, Looijenga LH. An oncofetal and developmental perspective on testicular germ cell cancer. Semin Cancer Biol. 2014.
- Burrell RA, Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol. 2014.
- Damia G, Garattini S. The pharmacological point of view of resistance to therapy in tumors. Cancer Treat Rev. 2014; 40: 909-916.
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013; 13: 714-726.
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002; 2: 48-58.
- Kramer RA, Zakher J, Kim G. Role of the glutathione redox cycle in acquired and de novo multidrug resistance. Science. 1988; 241: 694-697.
- Bandyopadhyay U, Das D, Banerjee R. Reactive oxygen species: Oxidative damage and pathogenesis. Current Science. 1999; 77: 658-666.
- Kerr JF. History of the events leading to the formulation of the apoptosis concept. Toxicology. 2002; 181-182: 471-4.
- Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxid Med Cell Longev. 2010; 3: 23-34.
- Ivanova D, Bakalova R, Lazarova D, Gadjeva V, Zhelev Z. The impact of reactive oxygen species on anticancer therapeutic strategies. Adv Clin Exp Med. 2013; 22: 899-908.
- Renschler MF. The emerging role of reactive oxygen species in cancer therapy. Eur J Cancer. 2004; 40: 1934-1940.
- Díaz-Laviada I, Rodríguez-Henche N. The potential antitumor effects of capsaicin. Prog Drug Res. 2014; 68: 181-208.
- Tian X, Yin H, Zhang S, Luo Y, Xu K, Ma P, et al. Bufalin loaded biotinylated chitosan nanoparticles: an efficient drug delivery system for targeted chemotherapy against breast carcinoma. Eur J Pharm Biopharm. 2014; 87: 445-453.
- Zhu X, Wang K, Zhang K, Zhu L, Zhou F. Ziyuglycoside II induces cell cycle arrest and apoptosis through activation of ROS/JNK pathway in human breast cancer cells. Toxicol Lett. 2014; 227: 65-73.
- Shen B, He PJ, Shao CL. Norcantharidin induced DU145 cell apoptosis through ROS-mediated mitochondrial dysfunction and energy depletion. PLoS One. 2013; 8: e84610.
- Izeradjene K, Douglas L, Tillman DM. Reactive oxygen species regulate caspase activation in tumor necrosis factor-related apoptosis-inducing ligand-resistant human colon carcinoma cell lines. Cancer Res. 2005; 65: 7436-7445.
- Jung EM, Lim JH, Lee TJ. Curcumin sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induce apoptosis through reactive oxygen species-mediated upregulation of death receptor 5(DR5). Carcinogenesis. 2005; 26: 1905-1913.
- Lee YJ, Lee GJ, Baek BJ, Heo SH, Won SY, Im JH, et al. Cadmium-induced up-regulation of aldo-keto reductase 1C3 expression in human nasal septum carcinoma RPMI-2650 cells: Involvement of reactive oxygen species and phosphatidylinositol 3-kinase/Akt. Environ Toxicol Pharmacol. 2011; 31: 469-478.
- Lee YJ, Jeong HY, Kim YB, Lee YJ, Won SY, Shim JH, et al. Reactive oxygen species and PI3K/Akt signaling play key roles in the induction of Nrf2-driven heme oxygenase-1 expression in sulforaphane-treated human mesothelioma MSTO-211H cells. Food Chem Toxicol. 2012; 50: 116-123.
- Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W, et al. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic Biol Med. 2011; 50: 624-632.
- Boldogh I, Roy G, Lee MS, Bacsi A, Hazra TK, Bhakat KK, et al. Reduced DNA double strand breaks in chlorambucil resistant cells are related to high DNA-PKcs activity and low oxidative stress. Toxicology. 2003; 193: 137-152.
- Chen J, Adikari M, Pallai R, Parekh HK, Simpkins H. Dihydrodiol dehydrogenases regulate the generation of reactive oxygen species and the development of cisplatin resistance in human ovarian carcinoma cells. Cancer Chemother Pharmacol. 2008; 61: 979-987.
- Suzuki-Karasaki Y, Suzuki-Karasaki M, Uchida M, Ochiai T. Depolarization Controls TRAIL-Sensitization and Tumor-Selective Killing of Cancer Cells: Crosstalk with ROS. Front Oncol. 2014; 4: 128.
- Ralph SJ, Rodríguez-Enríquez S, Neuzil J, Moreno-Sánchez R. Bioenergetic pathways in tumor mitochondria as targets for cancer therapy and the importance of the ROS-induced apoptotic trigger. Mol Aspects Med. 2010; 31: 29-59.
- Vaquero EC, Edderkaoui M, Pandol SJ, Gukovsky I, Gukovskaya AS. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem. 2004; 279: 34643-34654.
- Ibuki Y, Akaike M, Toyooka T, Mori T, Nakayama T, Goto R, et al. Hydrogen peroxide is critical for UV-induced apoptosis inhibition. Redox Rep. 2006; 11: 53-60.
- Takahashi M, Higuchi M, Matsuki H, Yoshita M, Ohsawa T, Oie M, et al. Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol Cell Biol. 2013; 33: 815-829.
- Azad N, Rojanasakul Y, Vallyathan V. Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J Toxicol Environ Health B Crit Rev. 2008; 11: 1-15.
- Prabhakaran K, Li L, Borowitz JL, Isom GE. Caspase inhibition switches the mode of cell death induced by cyanide by enhancing reactive oxygen species generation and PARP-1 activation. Toxicol Appl Pharmacol. 2004; 195: 194-202.
- Azad N, Iyer A, Vallyathan V, Wang L, Castranova V, Stehlik C, et al. Role of oxidative/nitrosative stress-mediated Bcl-2 regulation in apoptosis and malignant transformation. Ann N Y Acad Sci. 2010; 1203: 1-6.
- Panda PK, Mukhopadhyay S, Behera B, Bhol CS, Dey S, Das DN, et al. Antitumor effect of soybean lectin mediated through reactive oxygen species-dependent pathway. Life Sci. 2014; 111: 27-35.
- Gundala SR, Yang C, Mukkavilli R, Paranjpe R, Brahmbhatt M, Pannu V, et al. Hydroxychavicol, a betel leaf component, inhibits prostate cancer through ROS-driven DNA damage and apoptosis. Toxicol Appl Pharmacol. 2014; 280: 86-96.
- Wang J, Li JZ, Lu AX, Zhang KF, Li BJ. Anticancer effect of salidroside on A549 lung cancer cells through inhibition of oxidative stress and phospho-p38 expression. Oncol Lett. 2014; 7: 1159-1164.
- Cho SG, Woo SM, Ko SG. Butein suppresses breast cancer growth by reducing a production of intracellular reactive oxygen species. J Exp Clin Cancer Res. 2014; 33: 51.
- Yuko I, Rensuke G. Role of Redox Controls of Caspase Activities in Regulation of Cell Death. Current Enzyme Inhibition. 2005; 1: 281-285.
- Green DR. Death and NF-kappaB in T cell activation: life at the edge. Mol Cell. 2003; 11: 551-552.
- Hahnfeldt P, Panigrahy D, Hlatky L. Tumor development under angiogenic signaling: a dynamical theory of tumor growth, treatment response, and postvascular dormancy. Cancer Res. 1999; 59: 4770-4775.
- Spitz DR, Sim JE, Ridnour LA, Galoforo SS, Lee YJ. Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann N Y Acad Sci. 2000; 899: 349-362.
- Lloyd R Kelland. Targeting Established Tumor Vasculature: A Novel Approach to Cancer Treatment. Current Cancer Therapy Reviews. 2005; 1: 1-9.
- Lin X, Zhang F, Bradbury CM, Kaushal A, Li L, Spitz DR, et al. 2-Deoxy-D-glucose-induced cytotoxicity and radiosensitization in tumor cells is mediated via disruptions in thiol metabolism. Cancer Res. 2003; 63: 3413-3417.
- Ahmad IM, Abdalla MY, Aykin-Burns N, Simons AL, Oberley LW, Domann FE, et al. 2-Deoxyglucose combined with wild-type p53 overexpression enhances cytotoxicity in human prostate cancer cells via oxidative stress. Free Radic Biol Med. 2008; 44: 826-834.
- Simons AL, Mattson DM, Dornfeld K, Spitz DR. Glucose deprivation-induced metabolic oxidative stress and cancer therapy. J Cancer Res Ther. 2009; 5 Suppl 1: S2-6.
- Shutt DC, O'Dorisio MS, Aykin-Burns N, Spitz DR. 2-deoxy-D-glucose induces oxidative stress and cell killing in human neuroblastoma cells. Cancer Biol Ther. 2010; 9: 853-861.
- Vibhuti A, Muralidhar K, Dwarakanath BS. Differential cytotoxicity of the glycolytic inhibitor 2-deoxy-D-glucose in isogenic cell lines varying in their p53 status. J Cancer Res Ther. 2013; 9: 686-692.
- Ledoux S, Yang R, Friedlander G, Laouari D. Glucose depletion enhances P-glycoprotein expression in hepatoma cells: role of endoplasmic reticulum stress response. Cancer Res. 2003; 63: 7284-7290.
- Cheng SC, Zhou J, Xie Y. P-glycoprotein expression induced by glucose depletion enhanced the chemosensitivity in human hepatocellular carcinoma cell-lines. Cell Biol Int. 2005; 29: 269-275.
- Fogg VC, Lanning NJ, Mackeigan JP. Mitochondria in cancer: at the crossroads of life and death. Chin J Cancer. 2011; 30: 526-539.
- Kitajima Y, Miyazaki K. The Critical Impact of HIF-1a on Gastric Cancer Biology. Cancers (Basel). 2013; 5: 15-26.
- Jung SN, Yang WK, Kim J, Kim HS, Kim EJ, Yun H, et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis. 2008; 29: 713-721.
- Wartenberg M, Hoffmann E, Schwindt H, Grünheck F, Petros J, Arnold JR, et al. Reactive oxygen species-linked regulation of the multidrug resistance transporter P-glycoprotein in Nox-1 overexpressing prostate tumor spheroids. FEBS Lett. 2005; 579: 4541-4549.
- Qutub AA, Popel AS. Reactive oxygen species regulate hypoxia-inducible factor 1alpha differentially in cancer and ischemia. Mol Cell Biol. 2008; 28: 5106-5119.
- Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP, et al. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002; 62: 3387-3394.
- Wartenberg M, Ling FC, Müschen M, Klein F, Acker H, Gassmann M, et al. Regulation of the multidrug resistance transporter P-glycoprotein in multicellular tumor spheroids by hypoxia-inducible factor (HIF-1) and reactive oxygen species. FASEB J. 2003; 17: 503-505.
- Chen J, Ding Z, Peng Y, Pan F, Li J, Zou L, et al. HIF-1α inhibition reverses multidrug resistance in colon cancer cells via downregulation of MDR1/P-glycoprotein. PLoS One. 2014; 9: e98882.
- Glasauer A, Chandel NS. Targeting antioxidants for cancer therapy. Biochem Pharmacol. 2014.
- Wartenberg M, Richter M, Datchev A, Günther S, Milosevic N, Bekhite MM, et al. Glycolytic pyruvate regulates P-Glycoprotein expression in multicellular tumor spheroids via modulation of the intracellular redox state. J Cell Biochem. 2010; 109: 434-446.
- Hong H, Lu Y, Ji ZN, Liu GQ. Up-regulation of P-glycoprotein expression by glutathione depletion-induced oxidative stress in rat brain microvessel endothelial cells. J Neurochem. 2006; 98: 1465-1473.
- Li L, Xu J, Min T, Huang W. Up-regulation of P-glycoprotein expression by catalase via JNK activation in HepG2 cells. Redox Rep. 2006; 11: 173-178.
- Pandey V, Chaube B, Bhat MK. Hyperglycemia regulates MDR-1, drug accumulation and ROS levels causing increased toxicity of carboplatin and 5-fluorouracil in MCF-7 cells. J Cell Biochem. 2011; 112: 2942-2952.
- Lo YL. A potential daidzein derivative enhances cytotoxicity of epirubicin on human colon adenocarcinoma caco-2 cells. Int J Mol Sci. 2012; 14: 158-176.
- Terada Y, Ogura J, Tsujimoto T, Kuwayama K, Koizumi T, Sasaki S, et al. Intestinal P-glycoprotein expression is multimodally regulated by intestinal ischemia-reperfusion. J Pharm Pharm Sci. 2014; 17: 266-276.
- Duan R, Hu N, Liu HY, Li J, Guo HF, Liu C, et al. Biphasic regulation of P-glycoprotein function and expression by NO donors in Caco-2 cells. Acta Pharmacol Sin. 2012; 33: 767-774.
- Kanagasabai R, Krishnamurthy K, Druhan LJ, Ilangovan G. Forced expression of heat shock protein 27 (Hsp27) reverses P-glycoprotein (ABCB1)-mediated drug efflux and MDR1 gene expression in Adriamycin-resistant human breast cancer cells. J Biol Chem. 2011; 286: 33289-33300.
- Thévenod F, Friedmann JM, Katsen AD, Hauser IA. Up-regulation of multidrug resistance P-glycoprotein via nuclear factor-kappaB activation protects kidney proximal tubule cells from cadmium- and reactive oxygen species-induced apoptosis. J Biol Chem. 2000; 275: 1887-1896.
- Nwaozuzu OM, Sellers LA, Barrand MA. Signalling pathways influencing basal and H(2)O(2)-induced P-glycoprotein expression in endothelial cells derived from the blood-brain barrier. J Neurochem. 2003; 87: 1043-1051.
- Chen CC, Chu CB, Liu KJ, Huang CY, Chang JY, Pan WY, et al. Gene expression profiling for analysis acquired oxaliplatin resistant factors in human gastric carcinoma TSGH-S3 cells: the role of IL-6 signaling and Nrf2/AKR1C axis identification. Biochem Pharmacol. 2013; 86: 872-887.
- Deng HB, Adikari M, Parekh HK, Simpkins H. Ubiquitous induction of resistance to platinum drugs in human ovarian, cervical, germ-cell and lung carcinoma tumor cells overexpressing isoforms 1 and 2 of dihydrodiol dehydrogenase. Cancer Chemother Pharmacol. 2004; 54: 301-307.
- Chow KC, Lu MP, Wu MT. Expression of dihydrodiol dehydrogenase plays important roles in apoptosis- and drug-resistance of A431 squamous cell carcinoma. J Dermatol Sci. 2006; 41: 205-212.
- Chen YJ, Yuan CC, Chow KC, Wang PH, Lai CR, Yen MS, et al. Overexpression of dihydrodiol dehydrogenase is associated with cisplatin-based chemotherapy resistance in ovarian cancer patients. Gynecol Oncol. 2005; 97: 110-117.
- Matsunaga T, Hojo A, Yamane Y, Endo S, El-Kabbani O, Hara A, et al. Pathophysiological roles of aldo-keto reductases (AKR1C1 and AKR1C3) in development of cisplatin resistance in human colon cancers. Chem Biol Interact. 2013; 202: 234-242.
- Adeniji AO, Chen M, Penning TM. AKR1C3 as a target in castrate resistant prostate cancer. J Steroid Biochem Mol Biol. 2013; 137: 136-149.
- Chen J, Emara N, Solomides C, Parekh H, Simpkins H. Resistance to platinum-based chemotherapy in lung cancer cell lines. Cancer Chemother Pharmacol. 2010; 66: 1103-1111.
- Pallai R, Simpkins H, Chen J, Parekh HK. The CCAAT box binding transcription factor, nuclear factor-Y (NF-Y) regulates transcription of human aldo-keto reductase 1C1 (AKR1C1) gene. Gene. 2010; 459: 11-23.
- Sosa V, Moliné T, Somoza R, Paciucci R, Kondoh H, LLeonart ME. Oxidative stress and cancer: an overview. Ageing Res Rev. 2013; 12: 376-390.
- Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood. 2002; 99: 3505-3516.
- Son Y, Kim S, Chung HT, Pae HO. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013; 528: 27-48.
- Hales EC, Taub JW, Matherly LH. New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of γ-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell Signal. 2014; 26: 149-161.
- Lobry C, Oh P, Mansour MR, Look AT, Aifantis I. Notch signaling: switching an oncogene to a tumor suppressor. Blood. 2014; 123: 2451-2459.
- Jeong WS, Jun M, Kong AN. Nrf2: a potential molecular target for cancer chemoprevention by natural compounds. Antioxid Redox Signal. 2006; 8: 99-106.
- Gorre ME, Sawyers CL. Molecular mechanisms of resistance to STI571 in chronic myeloid leukemia. Curr Opin Hematol. 2002; 9: 303-307.
- Hall MD, Handley MD, Gottesman MM. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol Sci. 2009; 30: 546-556.
- Pluchino KM, Hall MD, Goldsborough AS, Callaghan R, Gottesman MM. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist Updat. 2012; 15: 98-105.
- Laberge RM, Ambadipudi R, Georges E. P-glycoprotein mediates the collateral sensitivity of multidrug resistant cells to steroid hormones. Biochem Biophys Res Commun. 2014; 447: 574-579.
- KrzyÅanowski D, Bartosz G, Grzelak A. Collateral sensitivity: ABCG2-overexpressing cells are more vulnerable to oxidative stress. Free Radic Biol Med. 2014.
- Lorendeau D, Dury L, Genoux-Bastide E, Lecerf-Schmidt F, Simões-Pires C, Carrupt PA. Collateral sensitivity of resistant MRP1-overexpressing cells to flavonoids and derivatives through GSH efflux. Biochem Pharmacol. 2014; 90: 235-245.
- Hall MD, Marshall TS, Kwit AD, Miller Jenkins LM, Dulcey AE, Madigan JP, et al. Inhibition of Glutathione Peroxidase Mediates the Collateral Sensitivity of Multidrug-resistant Cells to Tiopronin. J Biol Chem. 2014; 289: 21473-21489.
- Lu LY, Ou N, Lu QB. Antioxidant induces DNA damage, cell death and mutagenicity in human lung and skin normal cells. Sci Rep. 2013; 3: 3169.
- Nguyen J, Ma Y, Luo T, Bristow RG, Jaffray DA, Lu QB, et al. Direct observation of ultrafast-electron-transfer reactions unravels high effectiveness of reductive DNA damage. Proc Natl Acad Sci U S A. 2011; 108: 11778-11783.