
Case Presentation
Austin J Clin Pathol. 2017; 4(1): 1048.
Sotos Syndrome or Cerebral Gigantism: Case Report
Giovanni EM¹, Santos CC²*, Georgevich RG³, Noro Filho GA4, Caputo BV5, Andia Merlin RY6, da Silva TW7, Marques FCC8, Mesquita AMM9
¹Chairman, PhD, Professor, Integrated Clinic Discipline. Coordinator of Center for Studies and Special Service for Patients. Professor, Postgraduate Dentistry Courses, UNIP, São Paulo, SP, Brazil
4,5,9PhD, Associate Professor, UNIP, São Paulo, SP, Brazil; Member of Center for Studies and Special Service for Patients
2,6,8Associate Professor and PhD student, UNIP, São Paulo, SP, Brazil; member of Center for Studies and Special Service for Patients
3,7MD Student, UNIP, São Paulo, SP, Brazil; Member of Center for Studies and Special Service for Patients.
*Corresponding author: Santos CC, UNIP, São Paulo, Brazil
Received: March 28, 2017; Accepted: May 16, 2017; Published: May 26, 2017
Abstract
Soto’s syndrome was first described in 1964 by Juan Sotos. It is a rare disease, occurring in about 1 in 15,000 newborns, and exhibits variable expression in symptoms and severity. It is an autosomal dominant disease characterized by excessive growth during childhood, such as macrocephaly, hyper growth, typical facial dysmorphisms, distinctive facial gestalt. Patient JPS, female, 17 years old, dark skin, born normal. It presents speech difficulties, mental deficiency, large hands and long arms. The main facial skull features are evidenced by high and narrow skull with a dorsal pattern, hypertelorism, low nasal forehead, broad and prominent forehead. There is no treatment for cerebral gigantism. Treatment is symptomatic, but in general, pediatric follow-up is important during the first years of life to allow the detection and treatment of clinical complications such as scoliosis and febrile seizures. The early diagnosis of this syndrome is essential for a better neurological prognosis and for proper stimulation and referral to preventive dental treatment to be performed, and numerous sequel e caused by excessive head growth may be avoided.
Keywords: Gigantism; Soto’s syndrome; Mouth
Background and Review
Soto’s syndrome was first described in 1964 by Juan Sotos [1,2]. It is a rare disease, occurring in about 1 in 15,000 newborns, and exhibits variable expression in symptoms and severity (Figure 1). It is an autosomal dominant disease [3,4], characterized by excessive growth during childhood, such as macrocephaly, hypergrowth, typical facial dysmorphisms, distinctive facial gestalt (narrow and long face, pointed chin, prominent forehead, fine and sparse hair, hypertelorism with oblique slits down in eyelid) various degrees of learning difficulties due to mental deficiency and central nervous system disorders [5] (Figure 2). Many other clinical abnormalities may be present, such as: scoliosis, cardiac, genitourinary, renal, seizures, variable delays in cognitive and motor development [6,7]. Facial changes are more characteristic between 12 months and 6 years of age [8,9]. Diagnosis is assessed after birth, because of height overness and cephalic circumference (OFC), advanced bone age, including neonatal complications, and eating difficulties, hypotonia and facial gestalt [10,11]. During the neonatal period, the therapies are mainly symptomatic, including phototherapy in case of jaundice, and in the treatment of feeding difficulties, gastroesophageal reflux, and in the detection and treatment of hypoglycemia. Teeth eruption may occur prematurely (around 3 months), with maxillary hypoplasia, prognathism of the mandible leading to progression to Angel’s class III with anterior overbite, often reaching a vertical distance of up to 7 mm [12]. These patients are mostly mouth breathers and present difficulties in chewing and swallowing [13,14] (Figure 3 and Figure 4).

Figure 1: Front view.
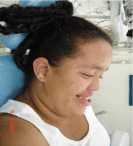
Figure 2: Side view.
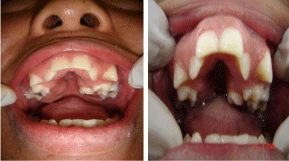
Figure 3 and 4: Regarding oral aspects: it presents ectopic teeth, oval palate.
NSD1 is the only known gene in association with Soto’s syndrome [15,16]. About 80-90% of individuals have a demonstrable mutation or deletion of NSD1, a gene that participates in normal development and growth [17]. Most mutations prevent a copy of the gene from encoding the protein or lead to the formation of a functionally abnormal and small protein [15,17,18]. It is not yet known how a reduced amount of NSD1 protein during development leads to overgrowth, learning difficulties and other signs and symptoms of Sotos Syndrome [7].
The syndrome may also be associated with an elevated risk of tumors [4,17,18]. Mutations and deletions of the NSD1 gene (located on chromosome 5q35 and encoding a histone methyltransferase involved in transcriptional regulation) account for more than 75% of cases [15]. Analysis of the MLPA or multiplex PCR quantitative allows the detection of total/partial NSD1 deletions, and direct sequencing allows the detection of NSD1 mutations [11]. The vast majority of NSD1 have abnormalities that occur more than once and there are very few familial cases. Although most cases are sporadic, several reports of autosomal dominant inheritance have been described [10,11].
The Genetic Test is indicated in the diagnosis, and the NGS-v1 Panel is indicated for the investigation of point mutations in NSD1, and of the MLPA-TCM MIC, which is the indicated test for duplication / deletion investigation of the region 5q35, which includes the NSD1 gene [5,6,13,15].
Sotos Syndrome makes differential diagnoses with several other pathologies, such as: Weaver’s syndrome, Beckwith-Wiedeman syndrome, Fragile-Golabi-Behmel X syndrome, Simpson syndrome, and 22qter deletion syndrome [2,14,15].
There is no treatment for cerebral gigantism [16]. Treatment is symptomatic, but in general, pediatric follow-up is important during the first years of life to allow the detection and treatment of clinical complications such as scoliosis and febrile seizures [17]. Educational psychologists who make adequate programs with speech therapy and motor stimulation play an important role in the overall development of patients, and the Dentist must be present from the diagnosis even in the inclusion and implementation of preventive measures appropriate to their needs, based on the degree of patient development, but overall treatment is completely multidisciplinary [13,16]. Final stature is difficult to predict, but growth tends to normalize after puberty [1,16].
Clinical Case
Patient JPS, female, 17 years old, dark skin, born normal. It presents speech and locomotion difficulties, mental deficiency, big hands, hypertelorism, prominent front, saddle nose, sparse hair on the anterior base of the head and on the eyebrows as well, discreetly inverted lips and long arms. The main facial cranial features are evidenced by high and narrow skull with dolic pattern, low nasal forehead, broad forehead and prominent. Still in the anamnesis, the mother reported being the third daughter in the marriage, with the first two children seemingly normal without evidence of alterations, and reported that the patient had premature loss of dental elements (Figure 5). We observed that she was mouth breather, had difficulties in swallowing and the diameter of her head was quite expressive, avoiding conventional patterns. Imaging was performed (panoramic x-ray), where we observed in the region of element 37 an image suggestive of condensing osteitis approximately 0.6 cm in diameter, clinically a gingival fibromatosis in the upper and lower elements, in addition to Angle Class III.

Figure 5: Panoramic X-ray - Ectopic teeth, decayed teeth, condensing
osteitis in the region of the element 37, and absence of the element 38.
The recommended clinical / therapeutic management was the performance of the salivary flow revealed around 0.8 ml / min, followed by the test of caries activity expressing the buffer capacity around 7.0, and evidence of bacterial plaque according to the Index O ‘Leary: 100%. A motivation protocol for oral hygiene technique was instituted, followed by prophylaxis in every extension of the oral cavity. The restorations with composite resin photoactivated in the elements: 12, 13, 14, 15, 24, 36, 45, 47, 48, 14, 17, 22, 24, 26. With the entire oral cavity recovered, within oral health standards, planning for gingivectomy was performed after orthodontic treatment.
Conclusion
The early diagnosis of this syndrome is essential for a better neurological prognosis and for adequate preventive stimulation to occur, facilitating the development of its functions. The Dentist has a primary role with the conduct of dental treatment being performed within the possibilities in a preventive way (Figure 6), which will avoid numerous sequels caused by excessive growth of the head. Sequelae that compromise the patient’s facial aesthetics. Multi and interdisciplinary involvement is extremely positive in the conduct of the treatments recommended by each specialty, becoming an important modifying factor in the promotion of health and in the improvement of patients’ quality of life.
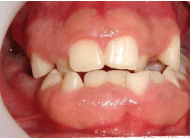
Figure 6: Gingival fibromatosis and Angle Class III.
References
- Bale AE, Drum MA, Parry DM, Mulvihill JJ. Familial Sotos syndrome (cerebral gigantism): craniofacial and psychological characteristics. Am J Med Genet. 1985; 20: 613-624.
- Baujat G, Rio M, Rossignol S, Sanlaville D, Lyonnet S, Le Merrer M, et al. Paradoxical NSD1 mutations in Beckwith-Wiedemann syndrome and 11p15 anomalies in Sotos syndrome. Am J Hum Genet. 2004; 74: 715-720.
- Cole TRP, Hughes HE. Sotos syndrome: a study of the diagnostic criteria and natural history. J Med Genet. 1994; 31: 20-32.
- Cole TRP, Hughes HE, Jeffreys MJ, Williams GT, Arnold MM. Small cell lung carcinoma in a patient with Soto’s syndrome: are genes at 3p21 involved in both conditions? J Med Genet. 1992; 29: 338-341.
- Douglas J, Coleman K, Tatton-Brown K, Hughes HE, Temple IK, Cole TRP, et al. The Childhood Overgrowth Collaboration. Evaluation of NSD2 and NSD3 in overgrowth syndromes. Europ J Hum Genet. 2005; 13: 150-153.
- Faivre L, Viot G, Prieur M, Turleau C, Gosset P, Romana S, et al. Apparent Sotos syndrome (cerebral gigantism) in a child with Trisomy 20p11.2-p12.1 mosaicism. Am J Med Genet. 2000; 91: 273-276.
- Hirai N, Matsune K, Ohashi H. Craniofacial and oral features of Soto’s syndrome: differences in patients with submicroscopic deletion and mutation of NSD1 gene. Am J Med Genet. 2011; 155: 2933-2939.
- Hoglund P, Kurotaki N, Kytola S, Miyake N, Somer M, Matsumoto N. Familial Sotos syndrome is caused by a novel 1 bp deletion of the NSD1 gene. J Med Genet. 2003; 40: 51-54.
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011; 13: 777-784.
- Kotilainen J, Pohjola P, Pirinen S, Arte S, Nieminen P. Premolar hypodontia is a common feature in Sotos syndrome with a mutation in the NSD1 gene. Am J Med Genet. 2009; 149: 2409-2414.
- Melchior L, Schwartz M, Duno M. dHPLC screening of the NSD1 gene identifies nine novel mutations--summary of the first 100 Sotos syndrome mutations. Ann Hum Genet. 2005; 69: 222-226.
- Sotos JF, Dodge PR, Muirhead D, Crawford JD, Talbot NB. Cerebral gigantism in childhood: a syndrome of excessively rapid growth with acromegalic features and a nonprogressive neurologic disorder. New Eng J Med. 1964; 271: 109-116.
- Fryssira H, Drossatou P, Sklavou R, Barambouti F, Manolaki N. Two cases of Soto’s syndrome with novel mutations of the NSD1 gene. Genet Couns. 2010; 21: 53-59.
- Mussa A, Chiesa N, Porta F, Baldassarre G, Silengo MC, Ferrero GB. The overlap between Sotos and Beckwith-Wiedemann syndromes. J Pediatr. 2010; 156: 1035-1036.
- Visser R, Landman EB, Goeman J, Wit JM, Karperien M. Sotos syndrome is associated with deregulation of the MAPK/ERK-signaling pathway. PLoS One. 2012; 7: e49229.
- Lane C, Milne E, Freeth M. Cognition and Behaviour in Sotos Syndrome: A Systematic Review PLoS One. 2016; 11.
- Park SH, Lee JE, Sohn YB, Ko JM, First Identified Korean Family with Sotos Syndrome Caused by a Novel Intragenic Mutation in NSD1. Ann Clin Lab Sci Spring. 2014; 44: 228-231.
- Berardi A, Quilici G, Spiliotopoulos, Corral Rodriguez MA, Martin Garcia F, Degano M, et al. Structural basis for PHDVC5HCHNSD1–C2HRNizp1 interaction: implications for Sotos syndrome. Nucleic Acids Res. 2016; 44: 3448-4346.