
Review Article
Austin J Clin Pathol. 2019; 6(1): 1058.
Role of Apoptosis in Human Diseases
Bautista-García PB¹*, García-Esquivel N², González-López L³, Vega-Campos I4, Medina- Barragán RA4, Medina-Sánchez MJ4, Salas- Medina DL4, Chávez-Estrada NE4, Montero- Castillo AB4, Rivera-Pérez ME4
¹School of High Studies in Health, La Salle University, Mexico
²National Autonomous University of México, School of Medicine. Campus Iztacala, State of Mexico, Mexico
³National Autonomous University of Mexico, School of Medicine. Mexico City, Mexico
4Autonomous University of Nayarit, Nursing Academic Unit, Mexico
*Corresponding author: Bautista-García PB, School of High Studies in Health, La Salle University, Camino Santa Teresa 811, Del. Tlalpan, ZIP 14018, Mexico City, Mexico
Received: December 31, 2018; Accepted: January 30, 2019; Published: February 06, 2019
Abstract
Apoptosis emerged as an accurate control of cell death, which is essential for development, differentiation, and homeostasis in organisms. However, under pathological conditions apoptosis can be related to disorders such as developmental abnormalities, cancer, autoimmune diseases, and pathophysiological effects in some organs. In this sense, programed cell death has been related to the onset and progression of many diseases including, cardiac failure, diabetes, liver and neurodegenerative diseases, which have a negative impact on the health system mainly in emerging economies. In this chapter, we will discuss the mechanisms involved in the onset and progression of apoptosis and its clinical impact in pathological conditions. In this way, the knowledge of the apoptosis mechanisms in health and pathology will be important in order to develop novel and accurate methods for its in vivo monitoring, including more sensible imaging techniques, identification of new serum biomarkers and DNA assays, which may estimate the level of cardiovascular, pancreatic, liver and central nervous system damage.
Keywords: Apoptosis; Cardiocyte apoptosis; ROS; Glucotoxicity; Lipoapoptosis; Amyloid beta
Introduction
In superior organisms apoptosis emerged as an accurate control of cells to maintain homeostasis during development and differentiation. In addition, apoptosis is a fundamental cellular process for removing unnecessary or potentially harmful cells by activating a geneticallyencoded suicide program within them. Although apoptosis is tightly regulated, in some instances, it can be misregulated causing disorders. In humans, these apoptosis-related disorders have been reported as developmental disorders, cancer, autoimmune diseases and pathophysiological effects, that can produce a negative impact on health. In the past, apoptosis was considered an autonomous process that did not affect surrounding cells, however recent studies had shown that apoptotic cells stablish an active communication with neighboring cells. The result of these cellular interactions includes enhancement of cell proliferation and morphogenetic changes of the neighbor cells due to mechanical stimuli [1].
Apoptosis signal pathways
Two types of programmed cell death have been described termed as intrinsic and extrinsic pathways, both exhibit specific features. The intrinsic pathway is induced by deleterious phenomena such as oxidative stress, radiation, intracellular pathogens, calcium overload, and DNA damage, leading to permeabilization of mitochondrial outer membrane by Bax/Bak pathway with a consequent translocation of cytochrome c from mitochondria to cytosol. Once in cytosol, cytochrome c and the Apaf-1 form the apoptosome which result in the caspase 9 activation.
Whereas, extrinsic apoptosis is incited by extracellular stress signals including binding of several cytokines such as, TNF-α, FasL, and TRAIL to their individual death receptors TNFR1. Death receptors then recruit FADD and procaspase 8 into the DISC, leading to caspase 8 activation. The activated initiator caspase 8 or 9 further induces activation of the effector caspases 3, 6, and 7, resulting in cleavage of essential cellular substrates and eventually apoptotic death [2-3] (Figure 1).
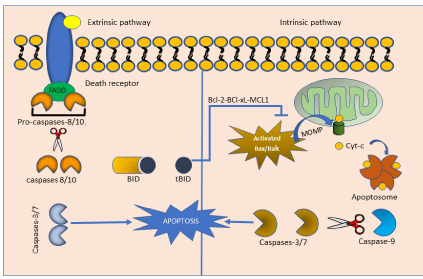
Figure 1: Intrinsic and extrinsic apoptosis pathway.
Although, apoptosis has been considered as a positive mechanism to control the proliferation ratio in a normal context its dysregulation also promotes several human diseases that will be reviewed in the next sections.
Cardiac myocyte apoptosis and its impact on human health
The heart functions as a pump in the cardiovascular system, which provide a continuous circulation of blood throughout the body. Apoptosis is a crucial process during the normal development of heart, however it has also been linked to an important number of cardiovascular pathological phenomena such as ischemic heart disease, ischemia-reperfusion injury, pharmacological induced cardiomyopathy, and heart failure [4].
In many cardiovascular diseases associated with failing myocardium such as left ventricular hypertrophy, the sustained activity of the renin-angiotensin-aldosterone system chronically increase intracellular calcium and the sympathetic response. Here, apoptosis plays a fundamental role as an early compensatory mechanism that can restore the cardiovascular homeostasis. However, as the pathological events progress, apoptosis also reduces significantly the normal function of the cardiovascular system [5-8].
Numerous mechanisms implicated in the induction of apoptosis in cardiopathy had been described, such as, the sustained activity of angiotensin II [9]. Related to this data, it has been showed that Angiotensin II activates proapoptotic p-53 transcription, which induces apoptogenic genes such as, cMyc, c-Fos, and c-Jun [10]. These data were supported by studies where the use of both, ACE inhibitors and Ang-II blockers prevent the harmful effects associated with the sustained activation of angiotensin II increasing afterload, ventricular remodeling, and cardiocytic and endothelial necrosis and apoptosis [11-12].
Calcium overload and cardiomyocytes apoptosis
Under physiological conditions calcium regulates several biological processes in heart including excitation-contraction coupling and cell survival and death. Intracellular calcium ions bind to the CaM in the cytosol and activate the Ca2+/CaM dependent phosphatase calcineurin and the CaMKII, leading to changes in cell physiology [13]. However, the persistent increase in intracellular calcium levels has been proposed as a harmful stimulus involved in myocardic apoptosis. The suppression of the STIM1: protein involved in intracellular Ca2+ accumulation by siRNA significantly decreased apoptosis and intracellular Ca2+ accumulation induced by hypoxia/ reperfusion H/R in H9C2 cardiomyocytes. In addition, suppression of STIM1 increase Bcl-2/Bax ratio and decrease Orai1/TRPC1 and caspase-3 activation [14].
Other mechanism proposed as a potent inductor of cardiomyocites cell death is the CaMKIIδ-mediated apoptosis which is associated NF-κB activation. In an Ischemia-Reperfusion [I/R] study in cardiomyocites, CaMKIIδ serves to the onset and sustained changes in the expression of proinflammatory genes which contribute to myocardial I/R damage and apoptosis [15].
Diabetic cardiomyopathy and apoptosis
During diabetes progression an important number of patients develop cardiovascular complications, such as DCM, which resulted in changes in the structure and function of the heart independently of other cardiac pathologies [16]. DCM is characterized by structural and functional changes including left ventricular hypertrophy and diastolic dysfunction. In this sense, diverse pathogenic mechanisms have been identified in DCM, such as myocardial cell death, contractile protein glycosylation and interstitial fibrosis. Myocardial cell apoptosis is a major component of DCM and it has been observed that these patients undergo dilated cardiomyopathy and showed more apoptotic cardiomyocytes than patients without diabetes [19] worsening their clinical evolution.
Additionally, it has been demonstrated that chronic hyperglycemia increases the level of ER stress and although it is initially considered an adaptive response to maintain ER homeostasis, the persistent activation can eventually trigger cell apoptosis and loss of function. Recent studies have shown that ER stress is an early event associated with diabetic cardiomyopathy triggered by hyperglycemia, FFAs and inflammation [18-19] (Figure 2).
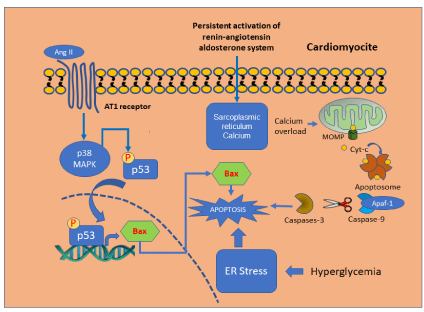
Figure 2: Mechanism involved in cardiomyocytes apoptosis.
Hepatocyte Apoptosis and its clinical impact on human health
Hepatocytes have a strong ability to regenerate when they are exposed to damaging factors or can undergone apoptosis in order to regulate homeostasis. However, under hepatic pathological conditions apoptosis can also contribute to both, chronic and acute liver diseases such as viral hepatitis, alcoholic and nonalcoholic liver disease, cholestatic disorders, and ischemia reperfusion injury diseases [20].
Hepatic apoptosis induced by Virus
Infection by hepatitis virus is one of the most important risk factors involved in the evolution of hepatic cirrhosis. Until now, it has been described five types of hepatitis virus, known as: hepatitis A, B, C, D, and E. However, only two families of virus are responsible of hepatic disease, Hepadnaviridae represented by the HBV and Flaviviridae by HBV.
For HBV, the Hepatitis B virus X protein (HBx) contributes to the progression of HBV-related hepatic injury and disease. In this case, HBx induce the overexpression of B56γ, a regulatory protein of the Protein Phosphatase 2A (PP2A), enhancing cell cycle arrest and inducing apoptosis. The mechanism related to hepatic cell death reside in the capability of AP1 to transactivate B56γ, which under the regulation of ER stress induce CREBH signaling in HBxexpressing hepatic cells. These cellular pathways promote that B56γbe dephosphorylated in p-Thr55-p53 triggering p53/p21 pathwaydependent cell cycle G1 phase arrest that result in apoptosis of hepatic cells [21].
Similarly, Zhang H et al. demonstrated that HBx is able to induce the expression of A20 through the upregulation of miR-125a, which in turn promotes TRAIL-induced hepatocyte apoptosis. A20 acts downstream of TNFR1 and counteract the apoptotic effect of TNF by the inactivation of the signaling molecule RIP1. In addition, HBx potentially inhibited the K63-linked ubiquitination of caspase-8 by downregulating A20 levels, enhancing the apoptosis cascade through caspase-8 cleavage, DISC formation and, hence, death signaling [22].
For HCV, its core protein and the structural protein (E1) can induce apoptosis. HCV core protein increase TRAIL-mediated apoptosis enhancing Bid cleavage and activating the mitochondria apoptosis signaling pathway. For the structural protein E1, it potentially alters membrane permeability through its hydrophobic transmembrane domain which causes apoptosis activation [23]. As mentioned above, these hepatitis virus have the capability to turn on the apoptosis cascade compromising the normal functioning of the liver and enhancing the accumulation of toxic substances which are potentially harmful to the patients.
Noninfectious liver disease and apoptosis
Although several mechanism of hepatocyte apoptosis have been recognized, other deleterious stimuli can potentially enhance programmed cell death. For example, hepatic steatosis induces notable hepatocyte apoptosis through several mechanisms. High-fat state is able to modify the metabolism to inhibit the activity of electron transport chain complexes and lengthen the life of electron transport intermediates, enhancing the production of ROS. In addition, high-fat diet can disrupt the tight-junction structure in intestinal epithelium and increase intestinal permeability, thus, enhancing the translocation of bacterial LPS into the liver activating Toll-like receptor 4 and increasing TNF-α secretion by the liver promoting hepatocyte apoptosis [24].
Alcoholic liver diseases
Another potential inductor of hepatic apoptosis is alcohol and its metabolites, both induce a significant production of ROS [25] with the subsequent decrease of mitochondrial GSH a potent antioxidant [26]. In addition, alcohol metabolites can also activate Fas/FasL and the downstream of the extrinsic apoptotic signaling pathway. Alcohol also induces ER stress in hepatocytes increasing mRNA levels of ER chaperones and caspase-12, which is a key element in ER stressrelated apoptosis [27].
Similarly, it has been shown that alcohol inhibits the expression of c-Met (a survival gene) through nuclear inactivation of the transcription factor Sp1 [28]. In addition, alcohol also participates in hepatic apoptosis elevating store-operated Ca2+ entry and leading to intracellular Ca2+ overload [29].
Drug-Induced hepatocyte apoptosis
Most drugs are metabolized by the liver and kidney; and due to their high metabolic activity, usually both are more susceptible to DI. A myriad of drugs can cause liver DILI through diverse pathways. DILI is induced by three different mechanisms: 1) direct hepatocyte injury; 2) exacerbation of a liver disease, especially hepatitis; and 3) a preexisting liver disease that alters drug pharmacokinetics by typically prolonging their effective time and enhancing their toxicity. Through liver metabolism, drugs can elicit electrophilic products. The chemical features of these metabolites will cause damage to the cell membrane as well as to the mitochondrial function and structure [30]. Another mechanism involved in the dose-dependent liver injury is the production of active metabolites, which exhaust cellular GSH and lead to lipid peroxidation and mitochondrial damage. ER stress is also believed to play a role in DILI [31].
Currently, the indiscriminate use of medical herbs has emerged as another mechanism for hepatic injury since herbs or infusions can be composed by potential toxic components such as, alkaloids, glycosides, and toxic proteins. The acute or chronic toxicity of these medical herbs may be associated with its pharmacokinetic and pharmacodynamics properties (doses, administration routes, lip solubility) and exist a strong correlation between long-term use and accumulative toxicity. For example, the herb TWHF is widely used in the Chinese traditional medicine; however, TP, the major active and toxic ingredient isolated from this herb is related with important side effects in several organs, including liver, kidney, testes, ovary, and heart in animals and even in humans. The toxic effects of TP are related to membrane damage, mitochondrial disruption, metabolism dysfunction, endoplasmic reticulum stress, oxidative stress, apoptosis and autophagy [32]. In this sense, the administration of herb, infusions or other compounds used for therapeutic purposes must be taken with caution because it is unknown the type and exact amount of the chemical compounds enhancing the potential toxicity (Figure 3).
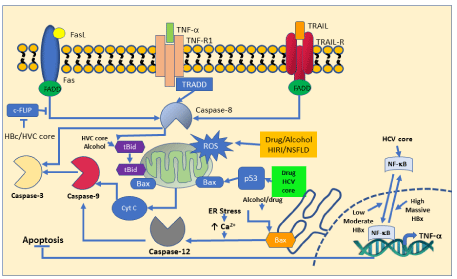
Figure 3: Mechanism involved in hepatocyte apoptosis.
Pancreatic Apoptosis and Its Clinical Impact
The pancreas is the organ involved in blood sugar control and its metabolism within the body. This organ is divided into an “endocrine” pancreas, which is related to the secretion of several biological mediators such as, insulin, glucagon, somatostatin and pancreatic peptide by the pancreatic islets helping controlling blood sugar levels and the metabolism within the body, and an “exocrine” pancreas, related to the secretion of enzymes involved in polysaccharides, proteins and fatty acids digestion. Both functions are involved in the normal function of the gastrointestinal tract.
Regarding the endocrine pancreas, approximately 3 million cell clusters called pancreatic islets have been quantified. These islets are composed by four main types of cells which are involved in the regulation of blood glucose levels. These cells produce an specific set of hormones: α (alpha) cells secrete glucagon, its main physiological role is to increase glucose concentration in blood, β (beta) cells secrete insulin, which enhance translocation of GLUT-4 into the plasma membrane decreasing glucose in blood, δ (delta) cells secrete somatostatin, which negatively regulates both synthesis and release of glucagon and insulin, and finally, PP cells, or γ (gamma) cells, which secrete pancreatic polypeptides [33-34]. Although, glucotoxicity might be the most important factor in the pathogenesis of diabetes another pathological harmful stimulus responsible of diabetes progression has been discovered. In the next section, processes involved in apoptosis of beta cells, especially secondary to increased glucotoxicity and lipotoxicity will be reviewed.
The Mechanism of Apoptosis in Diabetes
Although it is known that the main mechanism involved in the development of T1DM is the progressive loss of pancreatic beta cells by an erratic immune response, the mechanisms responsible for the appearance for T2DM are incompletely understand. In addition to the set of inflammatory cytokines, another organic compounds such as long-chain FFAs, ceramide, triacylglycerol, diacylglycerol, islet amyloid protein, and advanced glycation end products participate actively in the development of T2DM. In special, FFAs and its end products via JNK pathway play an important role in the pathogenesis of β-cell apoptosis (also named lipoapoptosis) [35].
During the progression of T2DM, other potential mechanisms in the pathogenesis of pancreas injury are involved including glucotoxicity and chronic inflammation which eventually increase oxidative stress and induce ER stress, that are closely related to β-cell dysfunction. These harmful phenomena might produce improper protein folding and the accumulation of these proteins could eventually cause β-cell ER stress and consequently beta-pancreatic cells death through the activation of CHOP (C/EBP homologous protein), caspase 12, JNK pathway, and IRE-1α which can trigger proapoptotic activity of Bcl-2, Bak, and Bax by PERK signaling pathway. In contrast, persistent ER stress can reduce ATF6 and IRE- 1α activity and induce PERK pathway. Indeed, the two main factors involved in the pathogenesis of diabetes, gluco and lipotoxicity, can participate in the onset and progression of beta-pancreatic cell death enhancing the Islet Amyloid Polypeptide (IAPP) synthesis, which under physiological conditions inhibits insulin and glucagon secretion. In contrast, in diabetic pancreas it has been demonstrated that IAPP can precipitate in aggregates (amyloid deposit), which can be inserted into the cell membrane of beta cells, where ion channel-like structures are formed. The pore forming capacity has been proposed to be an universal cytotoxic mechanism for all amyloid proteins. Furthermore, an aggregated IAPP can activate the inflammasome, a multiprotein oligomer responsible of the inflammatory response to produce processed interleukin-1 which may cause cell death [36-38]. Although, pharmaceutical agents have been developed to counteract the side effects of DM it is very important to know the mechanisms involved in the onset and progression of this pathology in order to develop novel medical and pharmaceutical strategies to prevent or ameliorate the appearance of DM especially in emerging countries where the health system is collapsed by the side consequences of diabetes (Figure 4).
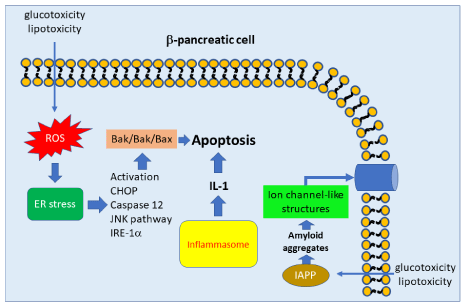
Figure 4: Mechanism involved in pancreatic apoptosis.
Neuronal Cell Death Mechanisms in Neurodegenerative Diseases
During the development of CNS, the active neurogenesis is often accompanied by massive neuronal loss [39] but once the entire programs of differentiation and development are completed, neuronal loss rarely occurs [40]. In contrast, in many ND, apoptosis is a response when harmful factors have surpassed the cell’s recovery capacity. In the following sections, the clinical impact of apoptosis during the onset and progression of ND will be discussed.
Alzheimer’s Disease (AD)
Memory loss, the most prominent clinical symptom in Alzheimer’s patients, is correlated with neuronal loss in the hippocampal region as well as in other areas of the CNS, such as entorhinal cortex layer II, the nucleus basalis of Meynert and the locus coeruleus. On this matter, Tau protein is implicated in the onset of AD. This protein under normal conditions is associated with the neuronal microtubules stabilizing its structure. Nevertheless, in neurodegenerative diseases, Tau is dissociated from neuronal microtubules forming aggregates Neurofibrillary Tangles (NFTs), which cannot be degraded by the proteasome system or the autophagic system resulting in cell death [41].
The relevant role of Tau protein in the pathogenesis of AD resides in its ability to interact with the pre-synaptic protein synaptogyrin-3, which may lead to defects in synaptic release, compromising neurons’ viability. Another potential mechanism of neural damage resides in the capability of Tau protein to stabilize NMDA receptor for its association with PSD-95 and Fyn. Tau may lead to overwhelming Ca2+ influxes through the same receptor, which leads to calpain activation and mitochondrial dysfunction, factors that cause cell death [42]. In addition, it has been also suggested that Tau leads to cell cycle re-entry and arrest at later stages increasing their susceptibility to cell death [43].
Parkinson’s Disease (PD)
The PD is characterized for a progressive loss of dopaminergic cells into the SN [44]. This loss result in an insufficient input of DA with a subsequent down-regulation of excitatory signals and upregulation of inhibitory signals in the motor loops circuit controlling body movement, leading to body movement-related symptoms such as rigidity and resting tremors in patients [45].
Many factors that contribute to the onset and progression of PD had been described. One of the most relevant factors is related to α-Synuclein, the major pathogenic protein of this pathology since its aggregates form the core of Lewy-bodies (which are alfa-synuclein aggregates), making nigral neurons more susceptible to the harmful effects of Lewy-bodies inducing a significant intrinsic ROS/RNS production and eventually cell death [46].
Although, it is not described how α-synuclein might induce neuronal death in PD, it is probable that its molecular aggregates induce mitochondrial dysfunction and oxidative stress. In this sense, in an in vitro study the α-synuclein overexpression in dopaminergic cells make the cells more susceptible to apoptosis in the presence of a minimal level of toxins. Another factor related to the pathogenic mechanism of α-synuclein is the fact that its aggregates can either suppress the communication between mitochondria and the ER or potentially turn on the apoptosis pathway or α-synuclein aggregates can cause lysosome membrane ruptures upon entering cells through endocytosis and augmenting ROS levels, which can also lead to cell death (Figure 5).
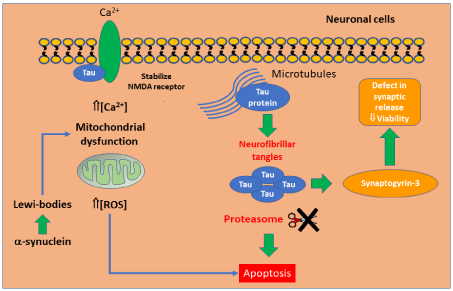
Figure 5: Mechanism involved in neuronal apoptosis.
Conclusion
Studies of the mechanism of cell death in the context of human diseases are of great value since they provide better strategies to prevent and retard the progression of cardiovascular, liver, neurodegenerative pathologies and diabetes. These pathologies produce negative effects in patient, families and health services especially in emergent economies. In the past decades, scientists have done important advances elucidating the apoptosis-related genes and signaling transduction pathways; however, no medical strategies exist to adjust the expression of apoptotic genes to prevent or alleviate some of the most common diseases in humans. Thereby it is indispensable develop new biomedical tools to identify the physiopathological changes associated with cell loss derived of several etiologic agents such as infectious agents, glucotoxicity, oxidative stress, drugs and ischemia-reperfusion phenomena to provide medical strategies to ameliorate, retard or prevent the functional disabilities associated several disease.
Conflict of Interest
No conflicts of interest, financial or otherwise, are declared by the author(s).
References
- Eroglu M, Derry WB. Your neighbours matter—Non-autonomous control of apoptosis in development and disease. Cell Death Differ. 2016; 23: 1110– 1118.
- Tixeira R, Poon IKH. Disassembly of dying cells in diverse organisms. Cell Mol Life Sci. 2018; 13.
- Pfeffer CM, Singh ATK. Apoptosis: A Target for Anticancer Therapy. Int J Mol Sci. 2018; 2: 19.
- Haunstetter A, Izumo S. “Apoptosis: basic mechanisms and implications for cardiovascular disease”. Circulation Research. 1998; 82: 1111–1129.
- Wang H, Zhang S, Xu S, Zhang L. The efficacy and mechanism of dexmedetomidine in myocardial apoptosis via the renin-angiotensinaldosterone system. J Renin Angiotensin Aldosterone Syst. 2015; 16: 1274- 1280.
- Marunouchi T, Tanonaka K. Cell Death in the Cardiac Myocyte. Biol Pharm Bull. 2015; 38: 1094-1097.
- Peng Xia, Yuening Liu, Zhaokang Cheng. Signaling Pathways in Cardiac Myocyte Apoptosis. BioMed Research International. 2016.
- Pérez-Schindler J, Philp A, Hernandez-Cascales J. Pathophysiological relevance of the cardiac β2-adrenergic receptor and its potential as a therapeutic target to improve cardiac function. Eur J Pharmacol. 2013; 698: 39-47.
- Zhao H, Qi G, Han Y, Shen X, Yao F, Xuan C, et al. 20-Hydroxyeicosatetraenoic Acid Is a Key Mediator of Angiotensin II-induced Apoptosis in Cardiac Myocytes. J Cardiovasc Pharmacol. 2015; 66: 86-95.
- Hernández JS, Barreto-Torres G, Kuznetsov AV, Khuchua Z, Javadov S. Crosstalk between AMPK activation and angiotensin II-induced hypertrophy in cardiomyocytes: the role of mitochondria. J Cell Mol Med. 2014; 18: 709- 720.
- Li X, Lan Y, Wang Y, Nie M, Lu Y, Zhao E.. Telmisartan suppresses cardiac hypertrophy by inhibiting cardiomyocyte apoptosis via the NFAT/ANP/BNP signaling pathway. Mol Med Rep. 2017; 15: 2574-2582.
- Chen Y, Yu S, Zhang N, Li Y, Chen S, Chang Y, et al. Atorvastatin prevents Angiotensin II induced myocardial hypertrophy in vitro via CCAAT/enhancerbinding protein β. Biochem Biophys Res Commun. 2017; 29: 423-430.
- Couchonnal LF, Anderson ME. The role of calmodulin kinase II in myocardial physiology and disease. Physiology (Bethesda). 2008; 23: 151-159.
- He F, Wu Q, Xu B, Wang X, Wu J, Huang L, et al. Suppression of Stim1 reduced intracellular calcium concentration and attenuated hypoxia/ reoxygenation induced apoptosis in H9C2 cells. Biosci Rep. 2017; 23: 37.
- Ling H, Gray CB, Zambon AC, Grimm M, Gu Y, Dalton N, et al. Ca2+/ Calmodulin-dependent protein kinase II δ mediates myocardial ischemia/ reperfusion injury through nuclear factor-κB. Circ Res. 2013; 15: 935-944.
- Asghar O, Al-Sunni A, Khavandi K, Khavandi A, Withers S, Greenstein A, et al. Diabetic cardiomyopathy. Clin Sci (Lond). 2009; 116: 741-760.
- Miki T, Yuda S, Kouzu H, Miura T. Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Fail Rev. 2013; 18: 149-166.
- Yang L, Zhao D, Ren J, Yang J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim Biophys Acta. 2015; 1852: 209-218.
- Lakshmanan AP, Harima M, Suzuki K, Soetikno V, Nagata M, Nakamura T, et al. The hyperglycemia stimulated myocardial Endoplasmic Reticulum (ER) stress contributes to diabetic cardiomyopathy in the transgenic non-obese type 2 diabetic rats: a differential role of Unfolded Protein Response (UPR) signaling proteins. Int J Biochem Cell Biol. 2013; 45: 438-447.
- Van Haele M, Roskams T. Hepatic Progenitor Cells: An Update. Gastroenterol Clin North Am. 2017; 46: 409-420.
- He C, Qiu Y, Han P, Chen Y, Zhang L, Yuan Q, et al. ER stress regulating protein phosphatase 2A-B56γ, targeted by hepatitis B virus X protein, induces cell cycle arrest and apoptosis of hepatocytes. Cell Death Dis. 2018; 9: 762.
- Ong B, Lv G, Wang Q, Wei F, Bellail AC, Hao C, et al. Targeting A20 enhances TRAIL-induced apoptosis in hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2012; 10: 433-438.
- Ciccaglione AR, Marcantonio C, Tritarelli E. The transmembrane domain of hepatitis C virus E1 glycoprotein induces cell death. Virus Res. 2004; 104: 1–9.
- Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol. 2014; 20: 14205–14218.
- Abdelmegeed MA, Choi Y, Ha SK, Song BJ. Cytochrome P450-2E1 promotes aging-related hepatic steatosis, apoptosis and fibrosis through increased nitroxidative stress. Free Radic Biol Med. 2016; 91: 188-202.
- Wu G, Yang J, Lv H, Jing W, Zhou J, Feng Y, et al. Taurine prevents ethanolinduced apoptosis mediated by mitochondrial or death receptor pathways in liver cells. Amino Acids. 2018; 50: 863-875.
- Liu LQ, Fan ZQ, Tang YF, Ke ZJ. The resveratrol attenuates ethanol-induced hepatocyte apoptosis via inhibiting ER-related caspase-12 activation and PDE activity in vitro. Alcohol Clin Exp Res. 2014; 38: 683-693.
- Tatsukawa H, Fukaya Y, Frampton G. Role of transglutaminase 2 in liver injury via cross-linking and silencing of transcription factor Sp1. Gastroenterology. 2009; 136: 1783–95e10.
- Cui R, Yan L, Luo Z, Guo X, Yan M. Blockade of store-operated calcium entry alleviates ethanol-induced hepatotoxicity via inhibiting apoptosis. Toxicol Appl Pharmacol. 2015; 287: 52–66.
- Kullak-Ublick GA, Andrade RJ, Merz M, End P, Benesic A, Gerbes AL, et al. Drug-induced liver injury: recent advances in diagnosis and risk assessment. Gut. 2017; 66: 1154-1164.
- Mosedale M, Watkins PB. Drug-induced liver injury: Advances in mechanistic understanding that will inform risk management. Clin Pharmacol Ther. 2017; 101: 469-480.
- Xi C, Peng S, Wu Z, Zhou Q, Zhou J. Toxicity of triptolide and the molecular mechanisms involved. Biomed Pharmacother. 2017; 90: 531-541.
- Morisset J. Seventy years of pancreatic physiology: take a look back. Pancreas. 2014; 43: 1172-1184.
- Saisho Y. Pancreas Volume and Fat Deposition in Diabetes and Normal Physiology: Consideration of the Interplay between Endocrine and Exocrine Pancreas. Rev Diabet Stud. 2016; 13: 132-147.
- Long J, Su YX, Deng HC. Lipoapoptosis pathways in pancreatic β-cells and the anti-apoptosis mechanisms of adiponectin. Horm Metab Res. 2014; 46: 722-727.
- Rojas J, Bermudez V, Palmar J, Martínez MS, Olivar LC, Nava M, et al. Pancreatic Beta Cell Death: Novel Potential Mechanisms in Diabetes Therapy. J Diabetes Res. 2018.
- Yao T, Fujimura T, Murayama K, Okumura K4, Seko Y. Oxidative Stress- Responsive Apoptosis Inducing Protein (ORAIP) Plays a Critical Role in High Glucose-Induced Apoptosis in Rat Cardiac Myocytes and Murine Pancreatic β-Cells. Cells. 2017; 18: 6.
- QiNan W, XiaGuang G, XiaoTian L, WuQuan D, Ling Z, Bing C. Par-4/NF- κB Mediates the Apoptosis of Islet β Cells Induced by Glucolipotoxicity. J Diabetes Res. 2016; 2016: 4692478.
- Kristiansen M, Ham J. Programmed cell death during neuronal development: the sympathetic neuron model. Cell Death Differ. 2014; 21: 1025-1035.
- Pfisterer U, Khodosevich K. Neuronal survival in the brain: neuron typespecific mechanisms. Cell Death Dis. 2017; 8: e2643.
- Nizynski B, Dzwolak W, Nieznanski K. Amyloidogenesis of Tau protein Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Protein Sci. 2017; 26: 2126-2150.
- Zhang Y, Li P, Feng J, Wu M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol Sci. 2016; 37: 1039-1047.
- Bougé AL, Parmentier ML. Tau excess impairs mitosis and kinesin-5 function, leading to aneuploidy and cell death. Dis Model Mech. 2016; 9: 307-319.
- Michel PP, Hirsch EC, Hunot S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron. 2016; 18: 675-691.
- Gopalakrishna A1, Alexander SA. Understanding Parkinson Disease: A Complex and Multifaceted Illness. J Neurosci Nurs. 2015; 47: 320-326.
- Chong W, Jiménez J, McIIvin M, Saito MA, Kwakye GF. α-Synuclein Enhances Cadmium Uptake and Neurotoxicity via Oxidative Stress and Caspase Activated Cell Death Mechanisms in a Dopaminergic Cell Model of Parkinson’s Disease. Neurotox Res. 2017; 32: 231-246.