
Review Article
Austin J Comput Biol Bioinform. 2016; 3(1): 1014.
Quantum-Chemical Description of the Propensity of Amino Acids of Formation of the Peptide Bond
Kereselidze J* and Mikuchadze G
Department of Chemistry, Ivane Javakhishvili Tbilisi State University, Georgia
*Corresponding author: Jumber Kereselidze, Department of Chemistry, Ivane Javakhishvili Tbilisi State University, 0179 Tbilisi, Georgia
Received: May 20, 2016; Accepted: June 16, 2016; Published: June 20, 2016
Abstract
With purpose of quantitative description of peptide bond formation the bond orders (PCO and PRNH), the bond lengths (RCO and RRNH), the charges on the carbon and nitrogen atoms (qC and qN) of carbonyl and amino groups, the activation energy (ΔE#) and the reaction Energy (ΔE) for 400 amino acid pairs by use the quantum - chemical method of Density Functional Theory (DFT) have been calculated. The formula of propensity of amino acids of peptide bonds formation (KP) by means these values were constructed.
Keywords: Amino acid; Peptide bond; Parameter of propensity; DFT calculations
Introduction
The theoretical description of biochemical processes for the development of the main direction of natural science - biophysical chemistry is very actual. In recent years, for a quantitative description of complex biochemical processes the modern method of quantum chemistry - Density Functional Theory (DFT) is widely used. Including and for investigation of the mechanism of peptide bond formation too [1]. It is assumed that the inductive and steric effects of the R-groups of amino acids have an effect on propensity of peptide bond formation [2]. A quantum-mechanical study of different possible mechanisms of peptide synthesis in the ribosome has been carried out using density functional also [3]. Analysis of database of protein sequences for all possible binary patterns of polar and non-polar amino acid residues revealed that alternating patterns occur significantly less often than others with similar composition. To facilitate understanding of the information available for protein structures, has been constructed the structural classification of proteins (scop) database. This database provides a detailed and comprehensive description of the structural and evolutionary relationships of the proteins of the known structure [4]. Analysis of extant proteomes has the potential of revealing how the frequencies of amino acids within proteins have evolved over biological time. It was shown that presented here residues of cysteine, tyrosine and phenylalanine have substantially increased in frequency [5]. To understand more fully how amino acid composition of proteins has changed over the course of evolution, a method has been developed for estimating the composition of proteins in an ancestral genome. This method was used to infer the amino acid composition of a large protein set in the Last Universal Ancestor (LUA) of all extant species. It is proposed that the inferred amino acid composition of proteins in the LUA probably reflects historical events in the establishment of the genetic code [6]. Several different formal definitions of local complexity and probability are presented and are compared for their utility in algorithms for localization of such regions in amino acid sequences and sequence databases. The occurrence of all di- and tripeptide segments of proteins was counted in a large data base containing about 119 000 residues. Systematic conformational analysis study of the tripeptidic units (Gly-X-Pro) and (Gly-Pro-X), with X = Pro, Ala, Ser, Val, Leu, Ile, and Phe it has been reported. The low-energy conformers obtained by quantum computations are discussed with respect to other theoretical investigations [7]. Model building revealed that of the 210 possible amino acid pairs of the standard 20 amino acids, no more than 26 could be built to meet standard criteria for bonding. Of these 26, 14 were found to be genetically encoded when the codons are read as if they paired in a parallel manner [8].
Methods
DFT is a computational quantum mechanical method and used in physical, chemical and biological sciences for investigate the electronic structure of molecules [9]. The properties of a manyelectron system can be determined by using functionals, which in this case is the spatially dependent electron density. Hence the name density functional theory comes from the use of functionals of the electron density. DFT is among the most popular and versatile methods available in computational biology. Hybrid methods, as the name suggests, attempt to incorporate some of the more useful features from ab initio methods (specifically Hartree-Fock methods) with some of the improvements of DFT mathematics. Hybrid methods, such as B3LYP [10-13], tend to be the most commonly used methods for computational chemistry and Biology.
Results and Discussion
The purpose of this paper is a quantitative description of the propensity of amino acids of formation of peptide bonds by means of quantum - chemical modern method of Density Functional Theory (DFT). All 400 options of pairing of the 20 amino acids are shown in (Table 1).
![]()
S No
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
1
GG
AG
SG
CG
VG
PG
TG
LG
AG
AG
GG
TG
TG
GG
AG
HG
MG
LG
IG
PG
2
GA
AA
SA
CA
VA
PA
TA
LA
AA
AA
GA
TA
TA
GA
AA
HA
MA
LA
IA
PA
3
GS
AS
SS
CS
VS
PS
TS
LS
AS
AS
GS
TS
TS
GS
AS
HS
MS
LS
IS
PS
4
GC
AC
SC
CC
VC
PC
TC
LC
AC
AC
GC
TC
TC
GC
AC
HC
MC
LC
IC
PC
5
GV
AV
SV
CV
VV
PV
TV
LV
AV
AV
GV
TV
TV
GV
AV
HV
MV
LV
IV
PV
6
GP
AP
SP
CP
VP
PP
TP
LP
AP
AP
GP
TP
TP
GP
AP
HP
MP
LP
IP
PP
7
GT
AT
ST
CT
VT
PT
TT
LT
AT
AT
GT
TT
TT
GT
AT
HT
MT
LT
IT
PT
8
GL
AL
SL
C L
VL
PL
TL
LL
AL
AL
GL
TL
TL
GL
AL
HL
ML
LL
IL
PL
9
GA
AA
SA
CA
VA
PA
TA
LA
AA
AA
GA
TA
TA
GA
AA
HA
MA
LA
IA
PA
10
GA
AA
SA
CA
VA
PA
TA
LA
AA
AA
GA
TA
TA
GA
AA
HA
MA
LA
IA
PA
11
GG
AG
SG
CG
VG
PG
TG
LG
AG
AG
GG
TG
TG
GG
AG
HG
MG
LG
IG
PG
12
GT
AT
ST
CT
VT
PT
TT
LT
AT
AT
GT
TT
TT
GT
AT
HT
MT
LT
IT
PT
13
GT
AT
ST
CT
VT
PT
TT
LT
AT
AT
GT
TT
TT
GT
AT
HT
MT
LT
IT
PT
14
GG
AG
SG
CG
VG
PG
TG
LG
AG
AG
GG
TG
TG
GG
AL
HL
ML
LL
IL
PL
15
GA
AA
SA
CA
VA
PA
TA
LA
AA
AA
GA
TA
TA
GA
AA
HA
MA
LA
IA
PA
16
GH
AH
SH
CH
VH
PH
TH
LH
AH
AH
GH
TH
TH
GH
AH
HH
MH
LH
IH
PH
17
GM
AM
SM
CM
VM
PM
TM
LM
AM
AM
GM
TM
TM
GM
AM
HM
MM
LM
IM
PM
18
GL
AL
SL
CL
VL
PL
TL
LL
AL
AL
GL
TL
TL
GL
AL
HL
ML
LL
IL
PL
19
GI
AI
SI
CI
VI
PI
TI
LI
AI
AI
GI
TI
TI
GI
AI
HI
MI
LI
II
PI
20
GP
AP
SP
CP
VP
PP
TP
LP
AP
AP
GP
TP
TP
GP
AP
HP
MP
LP
IP
PP
Table 1: All possible variants of pairing of amino acids.
For the study of the conformation of 20 amino acids by Kolaskar and Ramabraham 400 options were selected also [14]. The peptide bond can be formed in two ways: 1 - when the first amino acid is reacted by carboxy group and the second - with amino group and 2 - vice versa. They are sometimes referred to as peptides and antipeptides. Each column consists of a series, which is defined by the first letter of amino acid. For example, first column this is series of glycine. As is known, in the activation stage the peptide bond formation between two amino acids, the C-O bond of first amino acid and the N-H bond of the second amino acid is broken and with release of the water molecules and the C-N bond is formed (Figure 1). It has been suggested that, besides ribosome catalysis, the break these bonds is initiated by means of water molecules also [15]. With the aim of quantitative description of peptide bond formation by the quantum-chemical method DFT the bond orders of carbonyl and amino groups (PCO, PRNH) and lengths of corresponding bond (RCO, RNH), difference values of charge between C and N atoms (Δq), as well as the activation energy (ΔE#) and the reaction energy (ΔE) have been calculated. In addition, the exothermic nature of this process (ΔE<0) it was considered. The increase in bond lengths RCO and RNH, as well as the difference of charges between carbon atom of carbonyl group and nitrogen atom the amino group (Δq) causes an increase in the value of the parameter of the propensity of amino acids of peptide bond formation. This parameter we mark as Kp. These values in the formula Kp in the numerator are placed (Table 2). On the other hand, the decrease of bond orders PCO and the PRNH, as well as the activation energy ΔE# causes an increase in the parameter Kp. Hence, these values in the formula in denominator are located.
![]()
S No
Amino acid pairs
ΔE#, kJ/mol
ΔE, kJ/mol
qN
qC
Δq
PCO
PRNH
RCO, �
RRNH, �
KP .102
1
Ala-lya
52.5
-4.2
-0.135
0.175
0.31
0.94
0.76
1.45
1.1
1.32
2
Ala-Ala
65.6
17.6
-0.152
0.178
0.331
0.96
0.8
1.4
1.05
0.95
3
Ala-Ser
6.3
-44.6
-0.122
0.172
0.294
0.77
0.59
1.6
1.25
8.3
4
Ala-Cys
88.9
36.7
-0.165
0.171
0.336
0.89
0.8
1.4
1.05
0.79
5
Ala-Val
68.2
0.7
-0.14
0.181
0.321
0.93
0.77
1.45
1.1
1.06
6
Ala-Phe
55.1
3.4
-0.137
0.174
0.311
0.92
0.76
1.45
1.1
1.33
7
Ala-Thr
76.1
26.2
-0.162
0.178
0.34
0.95
0.81
1.4
1.05
0.85
8
Ala-Lys
78.7
26.1
-0.151
0.177
0.328
0.91
0.81
1.4
1.05
0.75
9
Ala-Asn
65.6
15.7
-0.139
0.177
0.316
0.92
0.76
1.45
1.1
1.07
10
Ala-Asp
56.4
2.1
-0.145
0.174
0.319
0.93
0.82
1.4
1.05
1.09
11
Ala-Gln
36.7
15.7
-0.136
0.179
0.315
0.92
0.76
1.45
1.1
1.52
12
Ala-Trp
57.8
3.9
-0.138
0.173
0.311
0.93
0.75
1.45
1.1
1.22
13
Ala-Tyr
82.2
28.9
-0.153
0.177
0.33
0.91
0.88
1.4
1.05
0.78
14
Ala-Glu
92.9
5.2
-0.129
0.172
0.301
0.86
0.77
1.45
1.1
0.79
15
Ala-Arg
76.1
14.2
-0.157
0.176
0.333
0.94
0.81
1.4
1.05
0.83
16
Ala-His
70.9
22.3
-0.148
0.176
0.324
0.97
0.81
1.4
1.05
0.84
17
Ala-Met
73.5
24.1
-0.143
0.178
0.321
0.97
0.81
1.4
1.05
0.81
18
Ala-Leu
52
1.3
-0.133
0.174
0.307
0.9
0.76
1.45
1.1
1.25
19
Ala-Ile
57.7
2.3
-0.135
0.174
0.309
0.92
0.76
1.45
1.1
1.22
20
Ala-Pro
60.4
7.9
-0.079
0.17
0.249
0.89
0.76
1.45
1.1
0.97
Table 2: Quantum-chemical characteristics for the alanine series.
Kp = Δq. RCO. RNH / ΔE#. PCO. PCO, ΔE<0
Among the 20 tables of 20 series, we present a table for series of alanine as example.
As the table shows the highest value of the Kp (8.3) observed for the ala-ser pair. This is in full accord with the conditions, according to which the formula of the parameter Kp was built. Besides the serinealanine pair in the series of serine is quite often meet [16]. It is known that the pairing of amino acids in proteins is encoded by sequence of nucleotide bases of the RNA. On other hand, the formation of peptide bonds is much dependent on the influence of inductive and steric effects of R-groups.
Waiting correlation between morphological data and the properties of R-groups seems hopeless, because under of their basis lie different concepts of natural science. The search of the correlations sometimes is thankless affair - better search the reasons of their absence. This may be the association of the R-groups with the ribosomal macromolecules or with water molecules. It should be noted that the energetically (?E#, ?E), electronic (qN, qC and ?q; PCO and PNH) and geometrically (RCO and RNH) characteristics contained in the table, has completely reasonable values, from the viewpoint of chemical notions.
All 20 series are shown in the list view that contain the amino acid pairs, for which Kp > 1, and they are located on symbating dependent curves Kp ~ Sij (Figure 2) and Kp ~ %, (Figure 3) where Sij frequency distribution of dipeptide [17] and % - calculated frequency of amino acids) [18]. Many of this pairs of amino acid cited in the Senger’s earlier works [19-21] and in [22] also. These amino acid pairs are boldered. As seen from (Figure 2) for 11 pairs from alanine series Kp symbatically depends from Sij. Hence, it can be assumed that our proposed the Kp parameter adequately describes the ability of the some (11) the amino acid pairs of a peptide bond formation. A similar relationship (Figure 3) from the calculated value of the amino acid sequency (%) is observed. Absence of complete adequacy of the parameter Kp can be explained by the different influence of reaction environment around the individual amino acids. However, must hope that proposed by us a quantum-chemical formula of parameter Kp can make a significant contribution of selection of the amino acid pairs for peptide bond formation.
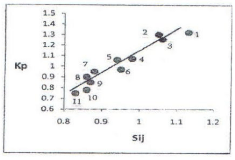
Figure 2: Dependency of the parameter of propensity of amino acids of
peptide bond formation (KP) from the normalized frequency distribution matrix
(SIJ) [9]. 1: ala-gly; 2: ala-gln; 3: ala-leu; 4: ala-asn; 5: ala-val; 6: ala-pro; 7:
ala-arg; 8: ala-ile; 9: ala-thr; 10: ala-tyr; 11: ala-lys.
The list contains pairs of amino acids 20 series, for which KP >1 and in earlier works Senger [19-21] and [22] are found also.
Given these conditions all amino acid pairs were distributed into three groups: low (1,1,2,2,3,3,3); middle (4,4,5,5,5,5,5) and the highest (6,6,7,7,8,8), where the numbers indicate the number of amino acid pairs in these series. From the suggested classification can draw some general conclusions. In particular the pairs of amino acids from series of tyrosine, leucine, lysine, glutamine, tryptophan and methionine should meet more often. Relatively less high frequency observed for the series of alanine, serine, aspartic acid, phenylalanine and glycine. Low frequency is characteristic for a series of isoleucine, asparagine, proline, tsisteyna, histidine, and threonine. From the individual amino acids the high frequency is characterized alanine (7), serine (9), glycine (10), lysine (11) and [23].
- seria (ala): ala-gly; ala-leu; ala-asn; ala-val. (4)
- seria (cys): cys-thr; cys-his. (2)
- seria (asp): asp-leu; asp-asp; asp-glu; asp-phe; asp-trp. (5)
- seria (glu): glu-ser; glu-cys; glu-gly; glu-val; glu-leu; gluasp; glu-trp. (7)
- seria (phe): phe-asp; phe-ser; phe-gly; phe-lys; phe-ile. (5)
- seria (gly): gly-glu; gly-leu; gly-val; gly-cys; gly-met. (5)
- seria (his): his-phe; his-his; his-asn. (3)
- seria (ile): ile-cys. (1)
- seria (lys): lys-his; lys-tyr; lys-gly; lys-ala; lys-ser; lys-leu; lys-pro. (7)
- seria (leu): leu-ile; leu-arg; leu-phe; leu-val; leu-ala; leuleu; leu-gly; leu-trp. (8)
- seria (met):met-his; met-tyr; met-pro; met-trp; met-ser; met-gln. (6)
- seria (asn): asn-lys. (1)
- seria (pro): pro-pro; pro-val. (2)
- seria (gln): gln-cys; gln-lys; gln-phe; gln-val; gln-gln. (5)
- seria (arg): arg-phe; arg-gln; arg-leu; arg-val; arg-gly. (5)
- seria (ser): ser-trp; ser-ser; ser-his; ser-ala. (4)
- seria (thr): thr-leu; thr-val; thr-pro. (3)
- seria (val): val-gln; val-trp; val-leu. (3)
- seria (trp): trp-ile; trp-met; trp-leu; trp-asn; trp-ser; trpthr. (6)
- seria (tyr): tyr-tyr; tyr-ser; tyr-pro; tyr-ile; tyr-met; tyrasn; tyr-gly; tyr-lys. (8) (Figure 1).
- "Kastner J, Sherwood P. The ribosome catalyzes peptide bond formation by providing high ionic strength. Molecular Physics. 2010; 108: 293-306.
- "Dwyer DS. Electronic properties of amino acid side chains: quantum mechanics calculation of substituent effects. BMC Chem Biol. 2005; 5: 2.
- "Acosta-Silva C, Bertran J, Branchadell V, Oliva A. Quantum-mechanical study on the mechanism of Peptide Bond. 2012; 134: 5817-5831.
- "Broome BM, Hecht MH. Nature disfavors sequences of alternating polar and non-polar amino acids: implications for amyloidogenesis. J Mol Biol. 2000; 296: 961-968.
- "Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: Structural Classification of proteins database. 2000; 28: 257-259.
- "Brooks DJ, Fresco JR, Lesk AM, Singh M. Evolution of amino acid frequencies in proteins over deep time: inferred order of introduction of amino acids into the genetic code. Mol Biol Evol. 2002; 19: 1645-1655.
- "Wootton JC, Federhen S. Statistics of local complexity in amino acid sequences and sequence database. Computer & Chemistry. 1993; 17: 149- 163.
- "Vonderviszt F, Matrai G, Simon I. Characteristic sequential residue environment of amino acids in proteins. Int J Pept Protein Res. 1986; 27: 483-492.
- "Cabrol D, Broch H, Vasilescu D. [Typical tripeptide sequences of collagen: quantitative conformation study]. Biochimie. 1981; 63: 851-855.
- "International Journal of Quantum Chemistry. Int J Quant Chem. 1983; 24: 109-122.
- "Root-Bernstein RS. Amino acid pairing. J Theor Biol. 1982; 94: 885-894.
- "Kohn W, Becke A, Parr R. Density Functional Theory of Electronic Structure. J Phys Chem. 1996; 100: 12974-12980.
- "Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A Gen Phys. 1988; 38: 3098-3100.
- "Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlationenergy formula into a functional of the electron density. Phys Rev B Condens Matter. 1988; 37: 785-789.
- "Perdew JP, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B Condens Matter. 1992; 45: 13244-13249.
- "Kolaskar AS, Ramabrahmam VR. Conformational properties of pairs of amino acids. Int J Pept Protein Res. 1983; 22: 83-91.
- "Wang Q, Gao J, Zhang D, Liu Ch. A theoretical model investigation of peptide bond formation involving two water molecules in ribosome support the twostep and eight membered ring mechanisms. Chemical Physics. 2015; 450- 451: 1-11.
- "Shen Sh, Bo Kai Bo, Ruan J, Huzil T, Carpenter E, Tuszynski JA. Probabilistic analysis of the frequencies of amino acid pairs within characterized protein sequences. Physical. 2006; 370: 651-662.
- "Senger F, Tuppy H. The amino acid Sequence in the Phenylalanyl Chain of Insulin. Biochemical Journal. 1951; 49: 481-490.
- "Senger F, Tompson EOP. The amino acid Sequence in the Glycyl Chain of Insulin. Biochemical Journal. 1953; 53: 353-366.
- "Baker HN, Gotto AM, Jackson RL. The primary structure of human plasma high density apolipoprotein glutamine I (ApoA-I). II. The amino acid sequence and alignment of cyanogen bromide fragments IV, III, and I. J Biol Chem. 1975; 250: 2725-2738.
- "Katti MV, Sami-Subbu R, Ranjekar PK, Gupta VS. Amino acid repeat patterns in protein sequences: their diversity and structural-functional implications. Protein Sci. 2000; 9: 1203-1209.
- "Atkins PW. The Second Law, Scientific Amerikan Books, an Imprint of W.H. Freeman and Company, New York. 1984.
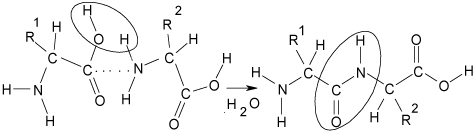
Figure 1: Scheme of the peptide bond formation resulting from the interaction
of different amino acid.
The symbatic dependence of the Kp from Sij is observed for only 11 pairs of amino acids from the series of alanine. Such selection of the amino acids pairs probably is caused by different direction of influence of inductive and steric effect of R-groups with respect to formation of peptide bonds. In other words, one R-group can promote other - prevent the formation of peptide bond. The dependence of Kp from the calculated frequency (%) [18] for the ten pairs of amino acids from different series is also a symbiotic (Figure 3). The similar situation can be expected from the remaining 19 series of amino acid pairs. From Figure 4 can be seen that the calculated frequency (%) of the nine amino acids symbatically depends on the charge of the carbon atom (qC) of the carboxy group. This relationship is consistent with the known phenomenon that in the step of initiating a amino acids aminoacyl adenylic acid on this carbon atom is acts. The higher
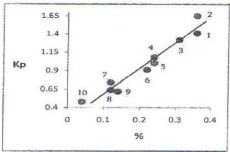
Figure 3: Dependency of the parameter of propensity of amino acids of
peptide bond formation (KP) from the calculated frequency (%) [18]. 1: argarg;
2: glu-ser; 3: pro-pro; 4: gln-gln; 5: glu-pro; 6: asn-asn; 7: his-pro; 8:
his-glu; 9: ile-met; 10: his-sis.
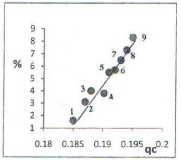
Figure 4: The dependency of the calculated frequency of amino acids (%) [9]
from the charge of carbon atom of carboxy group (qc). 1-Met; 2-Cys; 3- Asn;
4-Phe; 5- Asp; 6-Glu; 7-Val; 8-Ser; 9-Ala.
Conclusion
The proposed quantum-chemical formula, which includes the calculated values of energetic, electronic and structural characteristics of amino acids can be applied for quantitative description of the propensity of amino acids for the peptide bond formation as these values quantitatively describe inductive and steric influence of R-groups of amino acids on the reaction center during the peptide bond formation. These factors together with the coding by means nucleotide bases can have an important role in the study of the amino acid sequences in proteins. According to Atkins the orderliness can turn into chaos, and in certain conditions, from chaos it is possible the formation of the orderliness also [23].
Acknowledgment
This work was supported by the European Commission FP7, Progect High-perfomance Computing Infrastructure for South East Europe’s Research Communities, Grant No: 261499.
References