
Research Article
Austin Crit Care J. 2021; 8(1): 1036.
Histone Deacetylase Inhibitors as Potential COVID-19 Virus RNA-Dependent RNA Polymerase Inhibitors: A Molecular Docking and Dynamics Study
Mohamed MFA1*, Sayed AM2, Abdelmohsen UR3,4, Nafady A5, Khashaba PY6,7, Hayallah AM8*
1Department of Pharmaceutical Chemistry, Sohag University, Sohag, Egypt
2Department of Pharmacognosy, Nahda University, Beni-Suef, Egypt
3Department of Pharmacognosy, Minia University, Minia, Egypt
4Department of Pharmacognosy, Deraya University, Minia, Egypt
5Department of Chemistry, King Saud University, Riyadh, Saudi Arabia
6Department of Pharmaceutical Analytical chemistry, Assiut University, Assiut, Egypt
7Department of Pharmaceutical Chemistry, Sphinx University, New Assiut City, Egypt
8Department of Pharmaceutical Organic Chemistry, Assiut University, Assiut, Egypt
*Corresponding author: Alaa M. Hayallah, Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Sphinx University, New Assiut, Egypt, Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt
Mamdouh F.A. Mohamed, Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Sohag University, 82524- Sohag, Egypt
Received: March 30, 2021; Accepted: June 01, 2021; Published: June 08, 2021
Abstract
The novel coronavirus disease that initially appeared in 2019, commonly identified as COVID-19 is an acute infectious disease precipitated by the SARSCoV- 2 and has become a severe pandemic health crisis. People stricken with a severe case of COVID-19 are subject to a relatively higher mortality rate, which is brought about predominantly by the difficulty of having potent and specific antiviral drugs for its treatment. In this context, the viral RNA-dependent RNA polymerase (RdRp) is an attractive target for antiviral inhibitors, mainly because of its essential role in processing the polyproteins translated from viral RNA. Moreover, histone deacetylases inhibitors represent one of the most promising antiviral agents. Therefore, in this contribution, the in silico structure-based drug design approach was employed to identify novel structural characteristics for the potential repurposed activity of HDACIs as antivirals against COVID-19. Herein, 12 HDAC inhibitors were screened to explore their potential anti-viral activity against RNA-dependent RNA polymerase (RdRp) (6NUR). Results revealed that large number of the screened HDAC inhibitors are strongly bound into the active binding site of crystallographic structure of RNA-dependent RNA polymerase (RdRp) (6NUR) with lowest CDOCKER energy and CDOCKER interaction energy are very close to those of the control drug remdesivir. Importantly, the virtually screened HDAC inhibitors, particularly, Givinostat, Pracinostat, Panobinostat, Romidepsin (FK228) and its active metabolite (RedF) could be promising candidates for COVID-19 RNA-dependent RNA polymerase (RdRp) inhibitors. Significantly, these inhibitors should be evaluated on their effectivity when treating COVID-19, specifically using the drugs that have been approved for clinical trials with limited toxicity.
Keywords: COVID-19; HDAC inhibitors; Repurposing strategy; SARSCoV- 2; Molecular docking; dynamics
Introduction
Towards the end of year 2019, an unprecedented global outbreak of a newly identified coronavirus classified as severe acute respiratory syndrome (SARS-CoV-2), precipitated a global pandemic disease named COVID-19 [1–4]. The global health concerns have been elevated in recent times, due to the pandemic of (COVID-19) [5]. The extreme transmission rates and spread of disease have made the search for drug candidates to help in the reduction of this disease a global priority. A very successful strategy to combat a health crisis like this is drug repurposing, or more commonly recognized as repurposing or redirecting methods. Hence, drug redirecting of existing drugs is urgently required and could be a promising strategy for optimizing antiviral therapies that can successfully combat the SARS-CoV-2 infection in a short time. During the last few months, numerous FDA approved drugs and drugs under FDA examination have been repurposed for treating COVID-19 [6,7].
On the other hand, it is apparent that histone deacetylases inhibitors, in addition to their long history of use as targeted potent treatments for cancers [8,9], anti-epileptics, mood stabilizers [10]. antiparasitic [11] and anti-inflammatory [12], they have a potential role as novel therapeutic agents against viral infections [13]. The majority of HDAC inhibitors have three common pharmacophore patterns characterized as: A) Cap, B) linker, and C) Zinc Binding Group (ZBG) [14] as shown in Figure 1. Recently, six HDAC inhibitors (Figure 2) have been accepted as anticancer agents namely; Vorinostat (SAHA), [15] Romidepsin (FK228) and its active metabolite RedFK [16]. Belinostat (PXD101) [17], Pracinostat [18], Panobinostat (LBH-589) [19] and Chidamide (CS055) [20].
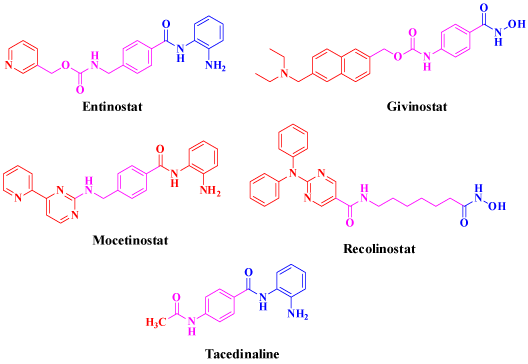
Figure 1: Structure of some anticancer HDACIs in clinical trials.
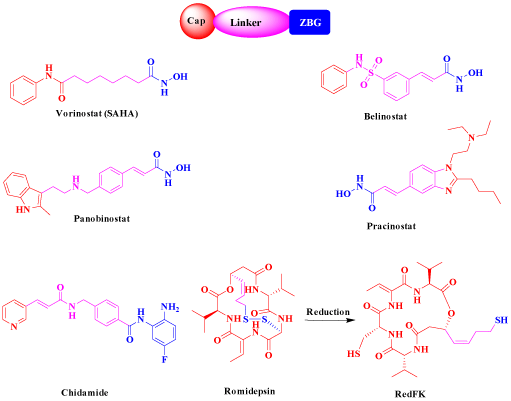
Figure 2: Structure of the approved anticancer HDACIs.
In addition, many other drugs are currently undergoing clinical trials to treat a diverse range of cancers such as Mocetinostat, Givinostat, Recolinostat, Tacedinaline, and Entinostat (Figure 1) [21–26].
It has been reported that Trichostatin A and valproic acid inhibited the appearance of inherent antiviral particles such as IFNβ, interferon-simulated genes, and other proteins involved in TLR3/TLR4 signaling. Additionally, they blocked microglial and astrocytic cytokine and chemokine gene expression, however, with different effects on other groups of cytokines [27,28]. Furthermore, TSA reduces the number of viral genomes in Herpes Simplex Virus-1 infected cells [29] (Figure 3).
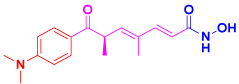
Figure 3: Trichostatin A.
Notably, in January 2013, after the successful initial round of in vitro research, the Danish Aarhus University Hospital was awarded $2 million to Dr. Ole Søgaard by the Danish Research Council, allowing them to proceed with clinical trials on 15 humans [30]. One report observed the use of vorinostat, entinostat, panobinostat, as well as romidepsin, precisely to reactivate latent HIV and mitigate the reservoirs. Vorinostat was stated as the lowest in its effectivity of the HDAC inhibitors within this trial [31]. Additionally, in another finding, it showed that romidepsin produced a higher and more persistent level of cell-associated HIV RNA reactivation than vorinostat in latently infected T-cells in vitro and ex vivo [32]. Furthermore, the hydroxamic acid derivative (BMY-26270) has been suggested it to be a selective inhibitor of purified influenza A RNAdependent RNA polymerase (RdRp) through IC50 equal to 40 μM. Also, it can inhibit influenza B in an equal potency; inhibited the invitro capped RNA-dependent transcription of the influenza B viral polymerase with equal potency [33,34] (Figure 4).
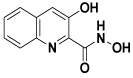
Figure 4: BMY-26270.
Within the search for potential drug targets, 3CLpro, RdRp, PLpro, and S, were the four functional proteins in 2019-nCoV that were examined as potential drug targets. Currently, there is no identified and approved antiviral drug for treatment of COVID-19. However, due to the impact it has had globally, there is a significant research push is now underway to repurpose existing drugs and to design new therapeutic agents targeting various components of the virus5. Evidently, the most efficient method to construct ant-2019- nCoV drug is to screen drugs which are currently being used in the clinic. RNA-dependent RNA polymerase (RdRp) is an imperative protease that catalyzes the replication of RNA from RNA template and is therefore an attractive therapeutic target for coronavirus. Hence, from the aforementioned effects of HDAC inhibitors on viral infections, it prompted us to further study and examine its ability to act as inhibiters for RNA-dependent RNA polymerase (RdRp) of COVID-19, via in silico method using molecular docking studies, with the aim of ultimately finding an effective treatment against COVID-19 infections.
Methods and Materials
Sequence alignment and modeling
Most of the promising clinical trials for COVID-19 treatments highlighted that the main antivirus drugs focus on proteins such as 3CLpro and RdRp which are much more preserved in SARS-COV and 2019-nCOV, specifically relating to its functionality. Therefore, our docking study focus on Rpdp as an important target. SARSCoV- 2 genes has been shown to share nucleotide identity and 89.10% nucleotide similarity with SARS-CoV genes in recent studies [35,36]. The SARS-HCoV resolved configuration (PDB ID: 6NUR) was used for binding, having a 97.08% sequence identity to SARS-CoV-2 RdRp. 6NUR is a SARS-HCoV non-structural protein 12 (nsp12) solved structure (cryo-electron microscopy) [37]. Moreover, Remdesivir was used as a control in this study, given it was the antagonist of 2019- COV RdRp.
Molecular docking methodology
Docking analysis was carried out by means of the Discovery Studio 2.5 software (Accelrys Inc., San Diego, CA, USA). Completely automatic docking tool using “Dock ligands (CDOCKER)”tive site with a standard RM procedure operating on Intel (R) core (TM) i32370 CPU @ 2.4 GHz 2.4 GHz, RAM Memory 2 GB under the Windows 7.0 system. Furthermore, these docked compounds were assembled using a software Chem 3D ultra 12.0 [Chemical Structure Drawing Standard; Cambridge Soft corporation, USA (2010)], and then sent to the Discovery Studio 2.5 software. From this, an automatic protein formulation procedure was conducted through the MMFF94 forcefield with the binding site sphere recognized by the software. The receptor was recorded as “input receptor molecule” in the CDOCKER protocol explorer. Establishing this, force fields were applied on the test compounds to obtain the minimum lowest energy structure. These poses were ranked and studied thoroughly, showing the best ligand-HDAC interactions from the calculations and 2D and 3D examinations [38- 41].
Molecular dynamic simulation
The Nanoscale Molecular Dynamics (NAMD) software was used in the molecular dynamic simulation (MDS) to understand the ligandenzyme complexes [42]. This was done by using the force field from the CHARMM27 [43]. Hydrogen atoms were added to the protein structures utilizing the psfgen plugin included Antibiotics 2020, 9, 562 14 of 16 in the Visual Molecular Dynamic (VMD) 1.9 software [44]. Subsequently, the entire framework was solvated using TIP3P water particles and 0.15M NaCl. This was carried out by minimizing the energy of the systems and then pprogressivel heating it up to 300 K and reach equilibrium for 200 seconds. Thereofore, the MDS was proceeded for 50 ns, and the trajectory was collected every 0.1 ns and further investigated with the VMD 1.9 software. These outputs were collected every 0.1 ns so that the conformational changes of the entire system can be considered using the Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF). From this, the VMD Force Field Toolkit (ffTK) [44] and an online software Ligand Reader & Modeler was used to examine the topologies and parameters of the compounds tested (http://www.charmm-gui.org/?doc=input/ ligandrm) [45].
Binding free energies were calculated through the free energy perturbation (FEP) technique, which was performed using the webbased software Absolute Ligand Binder [46].
Results and Discussion
Molecular modeling
RdRp: In order to investigate binding affinity between protein and the HDACIs, Discovery Studio software package was used. Twelve HDACIs were selected for the present study, the six approved HDACIs (SAHA, Romidepsin (FK228) and its active metabolite (RedF), Belinostat, Pracinostat, Panobinostat and Chidamide). In addition to five HDACIs in clinical trials, Recolinostat, Givinostat, Tacedinaline, Mocetinostat and Entinostat and comparing these results with the remdesivir.
The docking protocol was started by the docking of remdesivir in the SARS-HCoV solved structure (PDB ID: 6NUR, chain A). As shown in Figure 5, Table 1, remdesivir formed 6-H bonds with Gln444, Asn552, Asp445, Tyr455, Lys621 and Lys798, in addition to hydrophobic interaction with Ala554, Lys621 and Arg553. The CDOCKER energy of remdesivir is -30.1162 and the CDOCKER interaction energy is -59.1337.
![]()
CDOCKER interaction energy
No. of H-Bond
RedFK
-53.7436
8
Romidepsin
-35.9163
-52.8092
7
SAHA
-38.9654
-41.914
6
Belinostat
-28.8614
-43.0371
7
Pracinostat
-37.1318
-63.7588
6
Panobinostat
-45.8189
-50.0363
6
Chidamide
-33.2602
-44.6772
6
Tacedinaline
-32.2461
-40.437
5
Givinostat
-36.8399
-65.0395
7
Mocetinostat
-33.4505
-42.9723
5
Recolinostat
-36.001
-48.8773
5
Entinostat
-42.5925
-48.5569
6
Remdesivir
-30.1162
-59.1337
6
Table 1: CDOCKER energies and the energies produced from interactions, as well as the amount of hydrogen bonds of HDACIs into the active site of SARSHCoV solved structure (PDB ID: 6NUR).
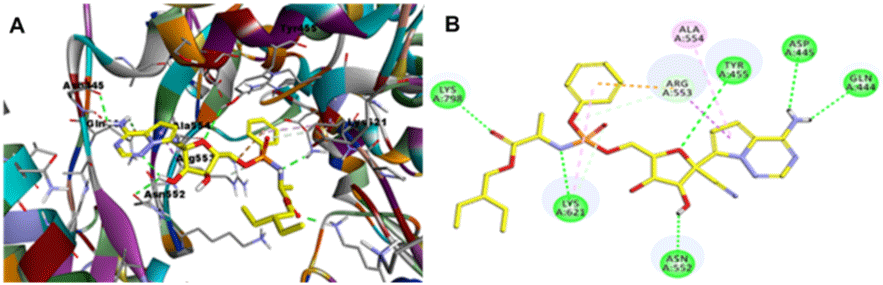
Figure 5: 3D (A) and 2D (B) The active site of SARS-CoV-2 RdRp within the binding stage of (PDB entry: 6NUR).
Among the twelve virtually screened HDAC inhibitors, five compounds; Romidepsin (FK228) and its active metabolite (RedF), Pracinostat, Panobinostat and Givinostat displayed CDOCKER energy and CDOCKER interaction energy better than the control drug remdesivir.
Givinostat, with -36.8399 CDOCKER energy and -65.0395 CDOCKER interaction energy, 7 hydrogen bonds were incorporated in the formation alongside amino acid remains Arg553 (2-H bonds), Thr556, Asp618, Asp623, Ser682 (2-H bonds). In addition to three strong attractive charges with Asp618, Asp760 and Glu811 and many other hydrophobic interactions with Asp618, Asp623, Arg624 and Asp760 (Figure 6).
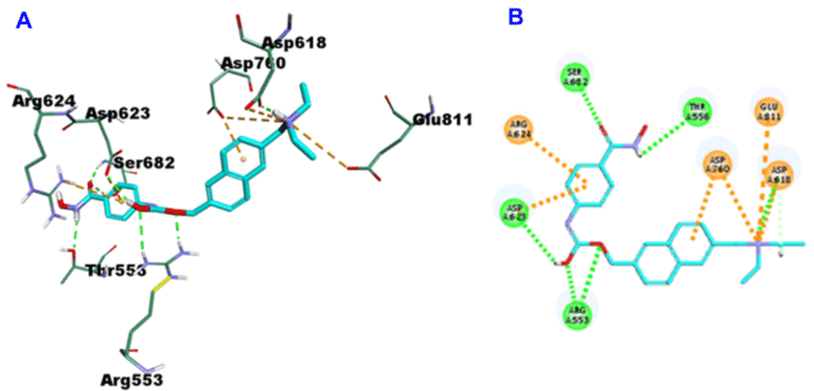
Figure 6: 3D (A) and 2D (B) binding mode of Givinostat into the active site of SARS-CoV-2 RdRp (PDB entry: 6NUR).
Regarding Pracinostat (Figure 7), with -37.1318 CDOCKER energy and -63.7588 CDOCKER interaction energy, it engaged in the formation of 6-H bonds with Lys545 (2-H bonds), Arg553 (2-H bonds), Arg555 and Tyr613 amino acid residues. In addition to eight hydrophobic interactions; two strong attractive charges with Asp618 and Asp760, two Pi-anion interactions with Asp623 and others with Tyr455, Tyr619, asp623, Asp618 and Asp760.
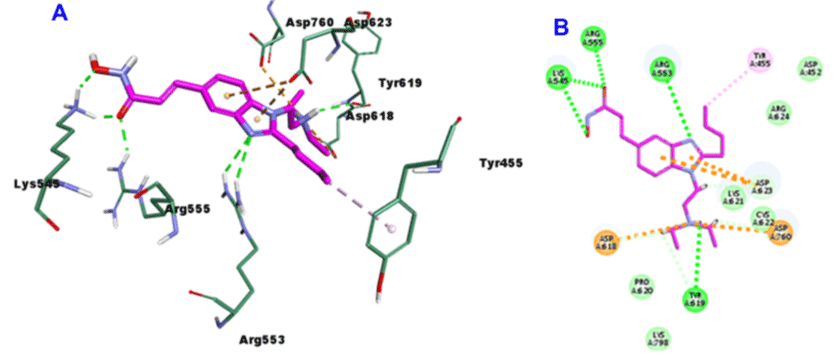
Figure 7: 3D (A) and 2D (B) binding mode of Pracinostat into the active site of SARS-CoV-2 RdRp (PDB entry: 6NUR).
As shown in Figure 8, Romidepsin active metabolite (RedF), with -40.3907CDOCKER energy and -53.7436 CDOCKER interaction energy, incorporated in the formation of 8-H bonds with the amino acid residues Arg553 (4-H bonds), Arg555, Thr556, Asp618 and Asp623, in addition to three hydrophobic interactions with Thr455, Lys545 and Arg624.
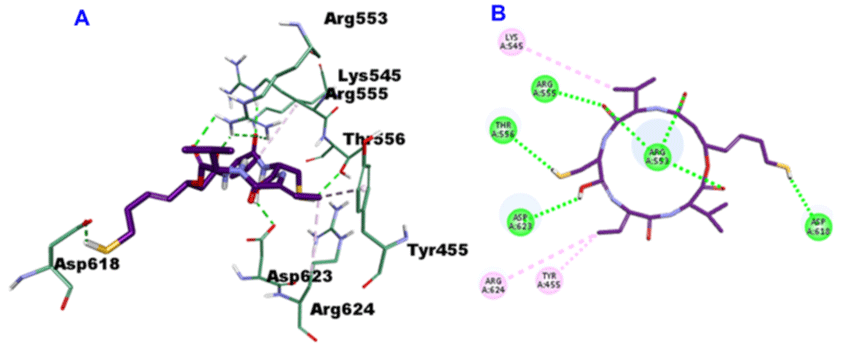
Figure 8: 3D (A) and 2D (B) binding mode of Romidepsin active metabolite (RedF) into the active site of SARS-CoV-2 RdRp (PDB entry: 6NUR).
Romidepsin, with -35.9163 CDOCKER energy and -52.8092 CDOCKER interaction energy, engaged in 7-H bonds with the amino acid residues Arg553 (3-H bonds), Lys551 (2-H bonds), Lys621 and Lys798, On top of three hydrophobic interactions alongside Aps618, Lys621 and Arg624 (Figure 9).
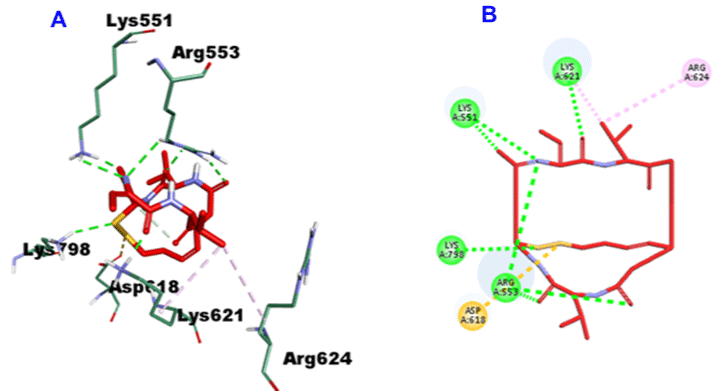
Figure 9: 3D (A) and 2D (B) binding mode of Romidepsin into the active site of SARS-CoV-2 RdRp (PDB entry: 6NUR).
Panobinostat, with -45.8189 CDOCKER energy and -50.0363 CDOCKER interaction energy, it involved in the formation of 6-H bonds with Lys545, Arg553 (2-H bonds), Arg555 (2-H bonds) and Asp623 amino acid remainders. On top of many hydrophobic interactions with Asp618, Lys621, Cys622 and two salt bridge interactions with Asp760 (Figure 10).
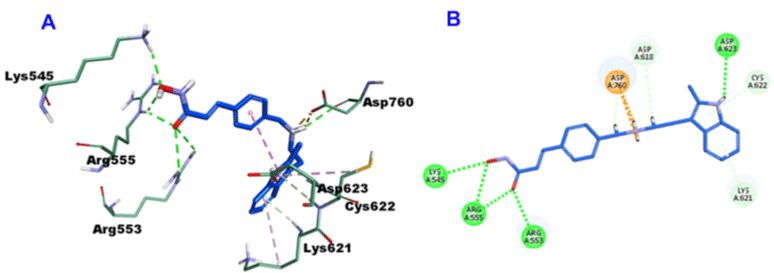
Figure 10: 3D (A) and 2D (B) binding mode of Panobinostat into the active site of SARS-CoV-2 RdRp (PDB entry: 6NUR).
Furthermore, all remaining virtually screened HDAC inhibitors; SAHA, Belinostat, Chidamide, Tacedinaline, Mocetinostat, Recolinostat, Entinostat, showed remarkable CDOCKER energy and CDOCKER interaction energy and involved in 5- or 6-H bonds as shown in Table 1 and Figure 11.
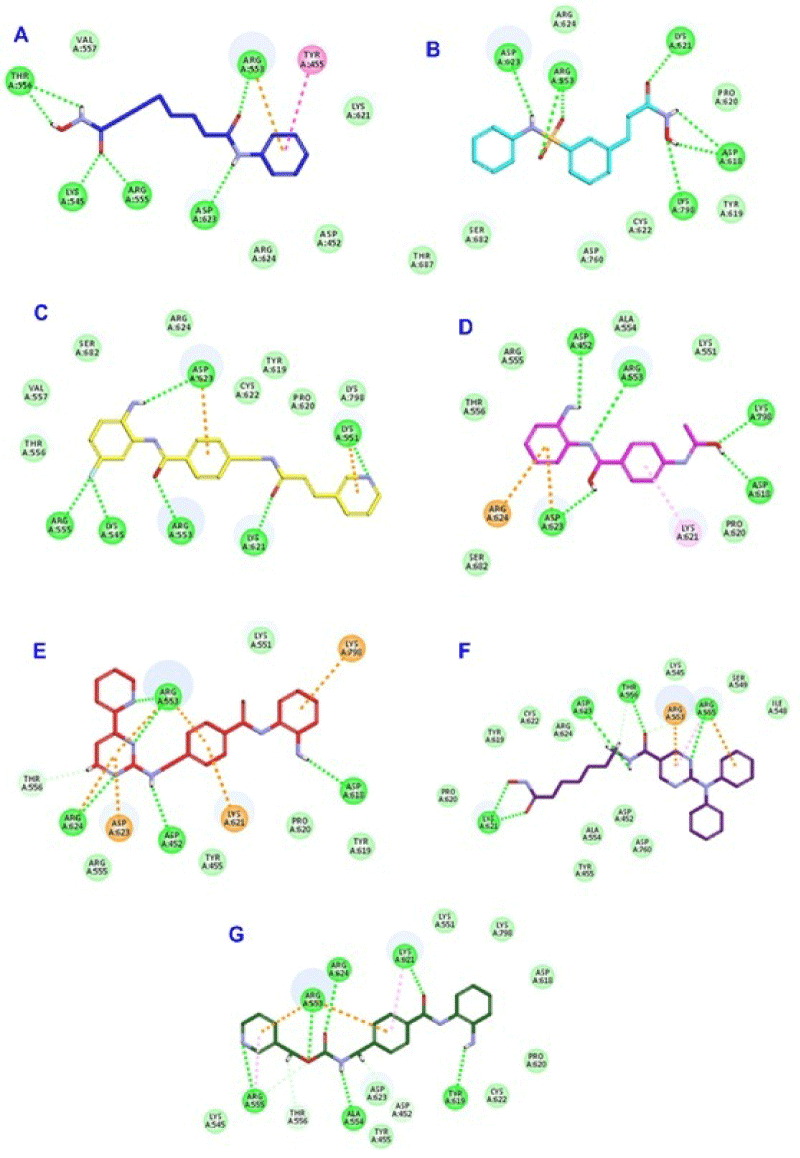
Figure 11: 2D binding mode of SAHA (A), Belinostat (B), Chidamide (C),
Tacedinaline (D), Mocetinostat (E), Recolinostat (F) and Entinostat (G) into
the active site of SARS-CoV-2 RdRp (PDB entry: 6NUR).
Molecular dynamic simulation analysis
To further validate our docking experiments, we subjected the protein-ligand complexes of the top-scoring compounds (Table 1) to a series of 50 ns Molecular Dynamic Simulations (MDS) (Figure 12). Givinostat achieved the highest binding stability inside the protein’s active site with a standard RMSD value of 1.8 Å, and plateaued at 14.2 ns. The next most stable drug was pracinostat (RMSD = 2.3 Å) which was able to achieve equilibrium (plateau) at 28.7 ns.
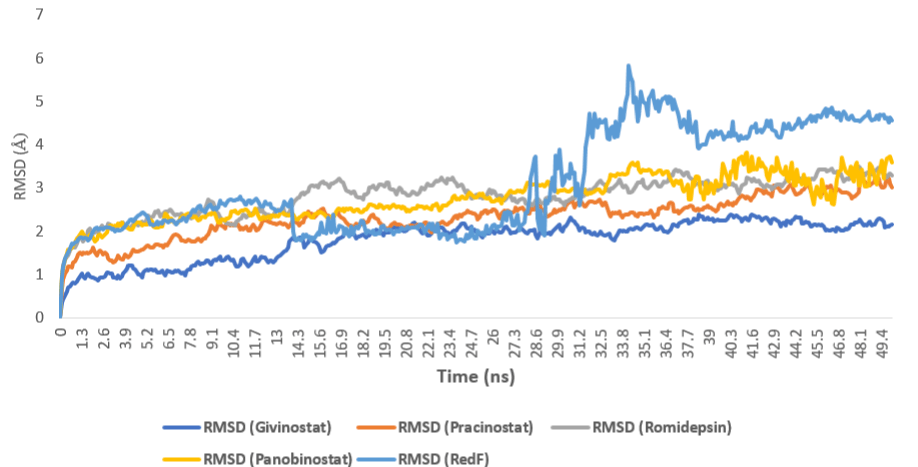
Figure 12: MDS-derived RMSDs of the top-scoring compounds inside the
protein’s active site over 50 ns.
Both romidepsin and panobinostat showed almost identical binding stability inside the protein’s active site (RMSD = 2.81 and 2.74 Å, respectively), where they reached the plateau at 24.7 ns.
Regarding the romidepsin’s metabolite RedF, its binding position was stable and similar to romidepsin and panobinostat till 13.8 (RMSD = 2.65 Å). Afterward, its RMSD decreased to about 2.1 Å till 27.7 ns. Starting from this point, the compound’s binding mode changed and its RMSD suddenly increased to reach its peak at 34.2 ns (RMSD = 5.84 Å). Subsequently, this transient increase of RMSD was stabilized at 4.2 Å from 38.4 ns till the end of the MDS. Hence, this compound was the least stable compound with an average RMSD of 3.1 Å.
Conclusion
In essence, the docking study revealed that the virtually screened approved HDAC inhibitors or in clinical trials of possible effective activity to be repurposed as COVID-19 SARS-CoV-2 RdRp inhibitors. This will encourage the examination of these drugs as anti-COVID-19, in particular, Givinostat, Pracinostat, Panobinostat, Romidepsin (FK228) and its active metabolite (RedF), as they bind tightly to the enzymes and displayed lowest CDOCKER energy and CDOCKER interaction energy better than or very close to the control drug remdesivir. This will be added to its HDAC inhibition activity. Hopefully, Givinostat, Pracinostat, Panobinostat, Romidepsin (FK228) and its active metabolite (RedF) could be promising candidates for COVID-19 RNA-dependent RNA polymerase (RdRp) inhibitors. Significantly, these inhibitors should be evaluated on their effectivity when treating COVID-19, specifically using the drugs that have been approved for clinical trials with limited toxicity.
Acknowledgment
This work was funded through Researchers Supporting Project number (RSP-2020/79) at King Saud University, Riyadh, Saudi Arabia.
References
- Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020; 395: 565–574.
- Li Q, Guan X, Wu P, et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N Engl J Med. 2020; 382: 1199– 1207.
- Zhou P, Yang X-L, Wang X-G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020; 579: 270–273.
- Wu F, Zhao S, Yu B, et al. Author Correction: A new coronavirus associated with human respiratory disease in China. Nature 2020; 580: E7–E7.
- Tahir ul Qamar M, Alqahtani SM, Alamri MA, et al. Structural basis of SARSCoV- 2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. 2020; 10: 313–319.
- Wu R, Wang L, Kuo H-CD, et al. An Update on Current Therapeutic Drugs Treating COVID-19. Curr Pharmacol Reports. 2020.
- El-Din Abuo-Rahma GA, Mohamed MFA, Ibrahim TS, et al. Potential repurposed SARS-CoV-2 (COVID-19) infection drugs. RSC Adv. 2020; 10: 26895–26916.
- Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: Overview and perspectives. Mol Cancer Res. 2007; 5: 981–989.
- Mwakwari SC, Patil V, Guerrant W, et al. Macrocyclic histone deacetylase inhibitors. Curr Top Med Chem. 2010; 10: 1423–1440.
- Slingerland M, Guchelaar H-J, Gelderblom H. Histone deacetylase inhibitors: an overview of the clinical studies in solid tumors. Anticancer Drugs. 2014.
- Patil V, Guerrant W, Chen PC, et al. Antimalarial and antileishmanial activities of histone deacetylase inhibitors with triazole-linked cap group. Bioorg Med Chem. 2010; 18: 415–425.
- Blanchard F, Chipoy C. Histone deacetylase inhibitors: new drugs for the treatment of inflammatory diseases? Drug Discov Today. 2005; 10: 197–204.
- Herbein G, Wendling D. Histone deacetylases in viral infections. Clin Epigenetics. 2010; 1: 13–24.
- Mohamed MFA, Youssif BGM, Shaykoon MSA, et al. Utilization of tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidinone as a cap moiety in design of novel histone deacetylase inhibitors. Bioorg Chem. 2019; 91: 103127.
- Kelly WK, O’Connor OA, Krug LM, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005; 23: 3923–3931.
- Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T, et al. FR901228, a novel antitumor bicyclic depsipeptide produced by chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J Antibiot (Tokyo). 1994; 47.
- Coiffier B, Pro B, Prince HM, et al. Results from a Pivotal, Open-Label, Phase II Study of Romidepsin in Relapsed or Refractory Peripheral T-Cell Lymphoma after Prior Systemic Therapy. J Clin Oncol. 2012; 30: 631–636.
- Novotny-Diermayr V, Hart S, Goh KC, et al. The oral HDAC inhibitor pracinostat (SB939) is efficacious and synergistic with the JAK2 inhibitor pacritinib (SB1518) in preclinical models of AML. Blood Cancer J. 2012; 2: e69–e69.
- Mottamal M, Zheng S, Huang TL, et al. Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules. 2015; 20: 3898–3941.
- Qiao Z, Ren S, Li W, et al. Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells. Biochem Biophys Res Commun. 2013; 434: 95–101.
- Santo L, Hideshima T, Kung AL, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012; 119: 2579– 2589.
- Gao S, Zang J, Gao Q, et al. Design, synthesis and anti-tumor activity study of novel histone deacetylase inhibitors containing isatin-based caps and o-phenylenediamine-based zinc binding groups. Bioorg Med Chem. 2017; 25: 2981–2994.
- Abdizadeh T, Kalani MR, Abnous K, et al. Design, synthesis and biological evaluation of novel coumarin-based benzamides as potent histone deacetylase inhibitors and anticancer agents. Eur J Med Chem. 2017; 132: 42–62.
- Xie R, Yao Y, Tang P, et al. Design, synthesis and biological evaluation of novel hydroxamates and 2-aminobenzamides as potent histone deacetylase inhibitors and antitumor agents. Eur J Med Chem. 2017; 134: 1–12.
- Trivedi P, Adhikari N, Amin SA, et al. Design, synthesis and biological screening of 2-aminobenzamides as selective HDAC3 inhibitors with promising anticancer effects. Eur J Pharm Sci. 2018; 124: 165–181.
- Li X, Zhang Y, Jiang Y, et al. Selective HDAC inhibitors with potent oral activity against leukemia and colorectal cancer: Design, structure-activity relationship and anti-tumor activity study. Eur J Med Chem. 2017; 134: 185–206.
- Suh H-S, Choi S, Khattar P, et al. Histone Deacetylase Inhibitors Suppress the Expression of Inflammatory and Innate Immune Response Genes in Human Microglia and Astrocytes. J Neuroimmune Pharmacol. 2010; 5: 521– 532.
- Dokmanovic M, Clarke C, Marks PA. Histone Deacetylase Inhibitors: Overview and Perspectives. Mol Cancer Res. 2007; 5: 981–989.
- Shapira L, Ralph M, Tomer E, et al. Histone Deacetylase Inhibitors Reduce the Number of Herpes Simplex Virus-1 Genomes Initiating Expression in Individual Cells. Front Microbiol. 2016; 7: 1970.
- Schwarzer R, Gramatica A, Greene WC. Reduce and Control: A Combinatorial Strategy for Achieving Sustained HIV Remissions in the Absence of Antiretroviral Therapy. Viruses. 2020.
- Elliott JH, Wightman F, Solomon A, et al. Activation of HIV Transcription with Short-Course Vorinostat in HIV-Infected Patients on Suppressive Antiretroviral Therapy. PLoS Pathog. 2014.
- Wei DG, Chiang V, Fyne E, et al. Histone Deacetylase Inhibitor Romidepsin Induces HIV Expression in CD4 T Cells from Patients on Suppressive Antiretroviral Therapy at Concentrations Achieved by Clinical Dosing. PLoS Pathog. 2014.
- De Clercq E. Antiviral agents active against influenza A viruses. Nat Rev Drug Discov. 2006; 5: 1015–1025.
- Cianci C, Chung TDY, Meanwell N, et al. Identification of N-Hydroxamic Acid and N-Hydroxyimide Compounds that Inhibit the Influenza Virus Polymerase. Antivir Chem Chemother. 1996; 7: 353–360.
- Zhou P, Yang X-L, Wang X-G, et al. Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin. bioRxiv. 2020.
- Tahir ul Qamar M, Alqahtani SM, Alamri MA, et al. Structural basis of SARSCoV- 2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. 2020; 10: 313–319.
- Yu R, Chen L, Lan R, et al. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int J Antimicrob Agents. 2020; 56: 106012.
- Abou-Zied HA, Youssif BGM, Mohamed MFA, et al. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg Chem. 2019; 89: 102997.
- Al-Sanea MM, Gotina L, Mohamed MFA, et al. Design, synthesis and biological evaluation of new HDAC1 and HDAC2 inhibitors endowed with ligustrazine as a novel cap moiety. Drug Des Devel Ther. 2020; 14: 497–508.
- Youssif BGM, Mohamed MFA, Al-Sanea MM, et al. Novel aryl carboximidamide and 3-aryl-1,2,4-oxadiazole analogues of naproxen as dual selective COX- 2/15-LOX inhibitors: Design, synthesis and docking studies. Bioorg Chem. 2019; 85: 577–584.
- Abd El-Kader AM, Abd El-Kader AM, Mahmoud BK, et al. Antiproliferative activity of new pentacyclic triterpene and a saponin from: Gladiolus segetum Ker-Gawl corms supported by molecular docking study. RSC Adv. 2020; 10: 22730–22741.
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005; 26: 1781–1802.
- MacKerell AD, Bashford D, Bellott M, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998; 102: 3586–3616.
- Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996; 14: 33–38.
- Jo S, Kim T, Iyer VG, et al. CHARMM-GUI: A web-based graphical user interface for CHARMM. J Comput Chem. 2008; 29: 1859–1865.
- Jo S, Jiang W, Lee HS, et al. CHARMM-GUI Ligand Binder for Absolute Binding Free Energy Calculations and Its Application. J Chem Inf Model. 2013; 53: 267–277.