
Special Article - Major Depressive Disorders
Ann Depress Anxiety. 2017; 4(1): 1085.
Clinical Impact of Pharmacogenetic Testing on Antidepressant Therapy
Holsboer F*
HMNC Holding GmbH, Germany
Corresponding author: Holsboer F, HMNC Holding GmbH, Maximilianstr. 34, 80539 Munich, Germany
Received: July 14, 2017;Accepted: September 01, 2017; Published: September 08, 2017
Editorial
Optimization of antidepressant treatment can be achieved by administration of gene tests that inform about pharmacokinetics. While the CYP450 gene test has merit in special cases the ABCB1-test informs about penetrance of a given drug into the brain. In both cases the prediction can be improved if combined with measurement of plasma drug concentrations. The implementation of gene tests targeting pharmacodynamics is not warranted until we know more about pathology of depression and antidepressants mode of action.
Keywords: Blood-Brain Barrier; CYP450 Enzymes; ABCB1 Test; Personalized depression treatment
Introduction
Current pharmacotherapy of depression follows a “trial-anderror” approach rather than a schedule where the first-line treatment is based on laboratory diagnostic that indicates the underlying pathology. Reasons that have fostered this sobering development include: (1) diagnostic categories are entirely based on mental symptoms; (2) patients fulfilling criteria for a certain category may have different disease mechanisms; and (3) in the absence of specific disease mechanisms „one-for-all“ drugs were developed that engage with multiple disease targets in the hopes that the disease causing mechanism is hit.
Clinicians tend to select their treatment according to past experience and according to specific side effects, e.g. sedation or activation, which may be useful in an individual case. Once a drug is selected, a treatment regimen is applied following the officially recommended guidelines. The adequacy of treatment and dosing is limited to what is called “therapeutic drug monitoring” to ensure that the administered drug dosage results in plasma drug concentrations that are within the appropriate range [1,2]. Prospective, controlled clinical trials where different patient groups were stratified according to fixed plasma drug levels and where clinical outcome is monitored to identify the perfect “therapeutic window” are yet not available. Instead, for each of the marketed antidepressants reference ranges were published that are largely based on expert consensus and not on studies. These ranges have considerable merit as guidance, but there is obviously room for further improvement.
In contrast to biomarkers that are physiologically and timely associated with the disease gene tests have the advantage of clinical feasibility as they are easily performed, independent from disease episodes and need no replication as gene variants are fixed. Thus, the main interest of all parties involved (patients, doctors and payers) currently focuses on gene tests [3].
Cytochrome P450 test
Cytochrome P450 proteins, predominantly located in the liver, have enzymatic properties and constitute the principal metabolic profile for most drugs in clinical use [4]. It is known from clinical observation and measurement of plasma drug levels that in some patients even moderate drug dosages result in high plasma concentrations while the same dosage in other patient’s results in very low drug levels. Obviously, some patients metabolize the administered drug very rapidly and are termed „ultra metabolizes“, while others having high plasma drug levels despite recommended dosages define as “poor metabolizes” [5]. Often also intermediate and extensive metabolizes between these extremes exist depending on the combination of CYP alleles they carry. As a result of different metabolize status “ultra metabolize” may be insufficiently dosed and therefore not responding to the drug while “poor metabolizes” may suffer from peripheral side effects secondary to the high plasma drug levels achieved by a standard dose.
Roche Diagnostics were the first to launch an oligonucleotide chip that measured genetic variations in two cytochrome p450 genes, CYP2D6 and CYP2C19, with limited commercial success. Today, the CYP2D6 and CYP2C19 genotyping is pursued by other companies, including STADA. While at first glance plausible, this test has some disadvantages as actual enzymatic activity is not precisely determined by gene polymorphisms. Enzyme activity, in general, is influenced by both, the substrate to be metabolized and the product that results from the chemical synthesis. More specifically, monotherapy among psychiatric patients is a rarity and some psychotropic drugs may impair CYP 450 enzyme activity while others can stimulate this activity. In pharmacokinetic studies administration of CYP2D6 inhibitors (e.g. paroxetine, fluoxetine) it was shown that individuals with non-poor metabolize genotype converted to a poor metabolizer phenotype. Larger studies, e.g. with venlafaxine, confirmed that phenoconversion to CYP2D6 poor metabolizer status among patients with non-poor metabolize genotype is occurring in around one quarter of patients [6]. Clearly, such phenotype conversions may have impact on clinical efficacy of a given drug. A prominent example where inhibition of a CYP 450 enzyme has considerable clinical implications is the inhibition of CYP1A2 by fluvoxamine that increases about threefold the plasma drug level of concomitantly administered clozapine which is also a CYP1A2 substrate [7]. There are other drugs that induce enzyme activity, e.g. carbamazepine, also smoking of tobacco induces CYP 450 enzyme activity.
In the light of these ambiguities treatment decisions guided by CYP 450 genotyping alone bears the risk of in proper dosing particularly if co medications are in place. In that case the individual patient is potentially either over- or under dosed. Such a risk can be avoided by continuous measurements of plasma drug levels which justify CYP genotyping only in special cases.
ABCB1 test
Endothelial cells of brain capillaries have tight junctions constituting the physical barrier whose paracellular transport of substances is negligible. Only passive diffusion, receptor-mediated transcytosis and specific carrier systems allow molecules to penetrate into the brain. Like all drugs used to treat brain disorders, including also antidepressants, regardless of clinical indication, must be able to penetrate to their site of action [8].
This biochemical efflux transporter, called ATP-Binding Cassette (ABC) transporters carries substrates across cell membranes including the endothelial cells of the blood-brain barrier. The phosphorylated glycoprotein, Pgp, is encoded by the ABCB1 gene and localized in the luminal (apical) cell membrane [9]. In mice where the ABCB1 gene is deleted the absence of Pgp results in enhanced neurotoxicity of various drugs supporting that ABCB1 expression is protecting the brain from exposure to potentially harmful lipophilic compounds that in the absence of the efflux transporter would diffuse into the central nervous system [10]. This applies only for antidepressants that are substrates of Pgp, i.e. they are bound at the Pgp binding domain, which induces a structural change of the Pgp molecule that leads to the drugs return transport into the blood circulation (Figure 1). Whether or not a given antidepressant is a Pgp substrate has been extensively studied using transgenic mice that carry an ABCB1 gene deletion. If the antidepressant concentration in the brain is much higher in ABCB1-knockout mice than in wild-type mice the antidepressant is a Pgp substrate. If the blood: brain ratio is unchanged by the absence of Pgp the drug is a non-substrate [11,12].
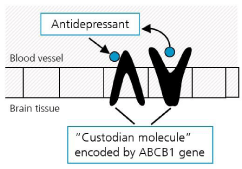
Figure 1: An antidepressant drug that is recognized as Pgp substrate is
pumped back by the efflux transporter Pgp that acts as “custodian molecule”.
In humans there exist a large number of genetic variants and studies interrogating if different ABCB1 variants translate into different clinical outcome. A total of 95 polymorphisms (SNPs) including exonic and intronic variants distributed over the whole ABCB1 gene were studied. Eleven intronic SNPs were associated with beneficial antidepressant treatment outcome, i.e. clinical remission. Two of these 11 intronic SNPs (rs 2032583, and rs 2235015) were found to be most informative. The key message was the following: In the presence of a certain gene variant, the C-allele of ABCB1 SNP rs 2032583, a significantly higher rate of patients that has achieved remission was found (Figure 2). Patients that carry the T-variant of the same SNP are less likely to remit under treatment with a Pgp substrate. Among patients treated with a non-substrate the ABCB1 genotype is irrelevant [13].
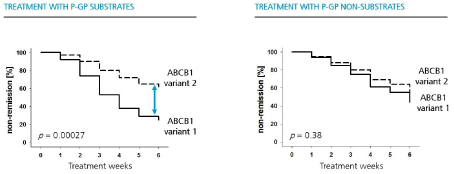
Figure 2: (adapted from Uhr et al. 2008): Patients that are treated with antidepressants that are Pgp substrates reach a remission of symptoms if the favourable
ABCB1-gene variant, the c-allele of the ABCB1 SNP rs2032583 was present (left). If treated with an antidepressant that does not bind to Pgp (non-substrate) the
ABCB1-genotype did not influence the treatment outcome (right).
This finding has prompted a number of replication studies using the originally reported variants and other predominantly exonic SNPs. The results led Breitenstein et al. to conduct a metaanalysis that focussed on prediction of clinical outcome by the most frequently investigated exonic (rs 1045642, rs 1128503, rs 2032582) and intronic (rs 2032583, rs 2235015, rs 2235040) SNPs [14]. Among the 16 studies included into the meta-analysis SNP rs 2032583 was the most informative: a nominal significant association across all studies was found for this SNP (p=0.035, in a total of 2037 patients) together with a significant Bonferroni correction among inpatients (p=1.5x10-5; 485 patients). Also SNP rs 2235015 was significantly (p=3.0x10-4) associated with treatment outcome withstanding Bonferroni correction in an inpatient subsample. All other SNPs were not informative.
The much better significance levels among inpatients highlights the effect of poor medication adherence among outpatients. This problem is well recognized as a general burden for clinical trials conducted in ambulatory settings and at different sites [15]. This is also the main reason for some mixed results from other studies that addressed ABCB1 testing. For example, the iSPOT-D trial, a multicenter study conducted in five different countries not taking into account ethnicity, drug dosage, and adjunctive treatments [16]. Unsurprisingly, that study failed to replicate the results by the same investigators obtained under more stringent study conditions. Clearly, people originating from different ethnic background may also show significant differences in DNA variability. This is one reason why the study by Dong et al. failed to corroborate the impact of both variants that Breitenstein and Uhr found [13,17,18]. Finally, the negative study by Perlis et al. points to an important issue that deserves more attention in future clinical applications [19]. This study used duloxetine as a prototypic substrate. In fact, definition of the substrate status does not follow an all or nothing principle, but is on a continuum between strong substrate and not a substrate at all. Duloxetine binds only weakly at Pgp and is therefore only a weak substrate. It needs to be considered to define a third category comprising weak substrates (Table 1). To investigate whether certain ABCB1-polymorphisms are predictive or not duloxetine is definitely an unsuited tool.
![]()
Antidepressants that are Pgp substrate
Examples for non-substrates
Paroxetine
Fluoxetine
Citalopram
Mirtazapine
Escitalopram
Agomelatine
Venlafaxine
Bupropione
Amitriptyline
Trazodone
Amitriptyline oxide
Lamotrigine
Nortriptyline
Trimipramine
Sertraline
Levomilnacipran
Vilazodone
Doxepine
Vortioxetine
Duloxetine (weak)
Hypericum
Table 1: List of Pgp substrates and non-substrates as determined using transgenic mice with and without functional genes encoding Pgp.
Another piece of evidence supporting the clinical benefit of ABCB1-testing is a comparison of clinical outcome among patients that were treated according to recommendations that came with the test result and patients that were not tested (Figure 3). Among tested inpatients the number of patients that achieved remission was significantly (p=0.005) higher than among untested inpatients [20].
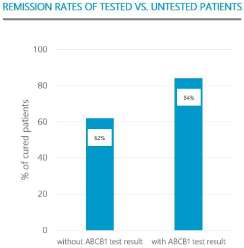
Figure 3: Patients were ABCB1 testing was administered and treatment was
adapted to the test result had a significantly (p = 0.005) better prospect of
remission.
In essence, ABCB1-testing across more than 32 published studies found the ABCB1-testing to be informative and resulting in a better clinical outcome or they failed to observe a benefit. The underlying methodological flaws underlying negative study results have been addressed above. Steering the treatment according to the ABCB1- test recommendations has not resulted in any negative clinical consequence. This has leaded the Swiss Society for Anxiety and Depression and the Swiss Society for Psychiatry and Psychotherapy to include ABCB1-testing into the official recommendations for treatment of acute depression [21].
It is of note that the functional role of ABCB1 gene variants upon Pgp-efflux activity is yet not resolved. To quantitative the impact of human ABCB1-SNPs upon drug concentrations in plasma and brain tissue must be measured which is not possible. Therefore, the conclusion that ABCB1-variants influence the penetrance of antidepressants into the brain is conjectural, but justified: Hoffmeyer et al. showed that the transport of Pgp substrates from the gut into the blood circulation is determined by ABCB1 gene variants [22]. Taken the clinical efficacy of antidepressants as surrogate of brain penetrance it is plausible that ABCB1 gene variants also influence passage of drugs across the blood brain barrier it is important to note that the relationship between ABCB1 gene variants and blood brain barrier passage are not carved in stone. Like other genes of that kind ABCB1 gene activity and Pgp expression are regulated by stressors and the excitatory neurotransmitter glutamate. In the light of current interest in glutamate targeting drugs the knowledge of ABCB1 gene variants may become even more important in clinical practice (Figure 4).
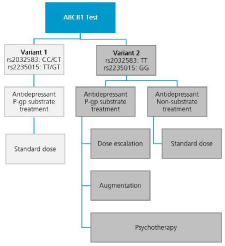
Figure 4: In case ABCB1-testing reveals the presence of the favourable gene
variant 1 (appr. 25% of all patients) treatment with a P-gp substrate (Table 1)
results in rapid response or remission. The only prerequisite is administration
of dosages and attainment of plasma drug levels as recommended by expert
panels. In the presence of the more frequently occurring (appr. 75% of all
patients) less beneficial variant of the ABCB1-gene poor treatment response
prompts consideration of the following options: (1) increase of dosage of
a P-gp substrate, (2) additional medication with a second antidepressant
and/or anticonvulsant or lithium (augmentation); and (3) treatment with a
non-substrate. In any case carrier with the frequent allele should receive
supportive psychotherapy to achieve optimal treatment outcome.
Multiplex gene test approaches
There are many reports highlighting singular genetic findings that associate with antidepressant treatment response. This raises the question if a multiplex gene test panel may be of benefit. A study by Winner et al. has recently reported a prospective, controlled trial that evaluates whether treatment of patients with major depression can be improved if a combinatorial five gene pharmacogenetic test is administered [23]. The test comprises Cyp 2D6, Cyp 2C19, Cyp 1A2 as pharmacokinetically relevant genes as well as the serotonin transporter gene SLC 6A4 and the serotonin 2A receptor gene HTR 2A representing pharmacodynamic mechanisms.
In that study one group received treatment as usual, the other group was treated according recommendations resulting from gene testing. Those patients that were tested and accordingly medicated had higher response (36% vs. 20.8%) and remission (20% vs. 8.3%) rates.
Outlook
The growing interest in laboratory diagnostics as guidance to improve antidepressant treatment is promising. While major innovations in antidepressant R & D are in the distant future clinicians will rely on better treatment modalities based on current antidepressants. The high numbers of patients which benefit incompletely or not at all are sobering. Any prospect to improve their situation should be welcomed not only by patients but also by doctors, payers, and regulatory offices.
Both, the CYP- and ABCB1-tests provide pharmacokinetic information while pharmacodynamic information is only included in the multiplex test. Our knowledge on mode of action of antidepressants is mute and no evidence exists that the mechanisms we know are those why they work. Therefore, the inclusion of pharmacodynamic gene polymorphisms as predictors of treatment remains arbitrary. Drug resistance, however, results from a complex interplay of pharmacokinetics and pharmacodynamics and particularly the latter are poorly understood. If a given drug does not engage with the disease mechanism it cannot work even if pharmacokinetics would support. On the other hand, if a drug would perfectly match with the disease causing mechanism it is essential that this drug reaches its target. The latter aspect can be improved with pharmacokinetic gene tests and brings the field a step closer to personalized depression therapy.
References
- Hiemke C, Baumann P, Bergemann N, Conca A, Dietmaier O, Egberts K, et al. AGNP Consensus Guidelines for Therapeutic Drug Monitoring in Psychiatry: Update 2011. Pharmacopsychiatry. 2011; 44: 195-235.
- Gründer G, Baumann P, Conca A, Zernig G, Hiemke C. Therapeutisches Drug-Monitoring in der Psychiatrie. Der Nervenarzt. 2014; 7: 847-855.
- Holsboer F. How can we realize the Promise of Personalized Antidepressant Medicines? Nat Rev Neurosci. 2008; 9: 638-646.
- Zanger UM, Schwab M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol Ther. 2013; 138: 103-141.
- Kirchheiner J, Brosen K, Dahl ML, Gram LF, Kasper S, Roots I, et al. CYP2D6 and CYP2C19 Genotype-based Dose Recommendations for Antidepressants: A First Step towards Subpopulation-Specific Dosages. Acta Psychiatr Scand. 2001; 104: 173-192.
- Preskorn S, Kane C, Lobello K, Nichols A, Fayyad R, Buckley G, et al. Cytochrome P450 2D6 Phenoconversion is Common in Patients Being Treated for Depression: Implications for Personalized Medicine. J Clin Psychiatry. 2013; 74: 614-621.
- Lu ML, Lane HY, Chen KP, Jann MW, Su MH, Chang WH. Fluvoxamine reduces the Clozapine Dosage needed in Refractory Schizophrenic Patients. J Clin Psychiatry. 2000; 61: 594-599.
- Chow B, Gu C. The Molecular Constituents of the Blood-Brain Barrier. Trends Neurosci. 2015; 38: 398-608.
- Cordon-Cardo C, O’Brien J, Casal D, Rittman-Grauer L, Biedler J, Melamed M, et al. Multidrug-Resistance Gene (P-Glycoprotein) is expressed by Endothelial Cells at Blood-Brain Barrier Sites. Proc Natl Acad Science USA. 1989; 86: 695-698.
- O’Brien F, Dinan T, Griffin B, Cryan J. Interactions between Antidepressants and P-Glycoprotein at the Blood-Brain Barrier: Clinical Significance of in vitro and in vivo Findings. Br J Pharmacol. 2012; 165: 289-312.
- Doran A, Obach R, Smith B, Hosea N, Becker S, Callegari E, et al. The Impact of P-Glycoprotein on the Disposition of Drugs targeted for Indications of the Central Nervous System: Evaluation using the MDR1A/1B Knockout Mouse Model, Drug Metab Dispos. 2005; 33: 165-174.
- Uhr M, Steckler T, Yassouridis A, Holsboer F. Penetration of Amitriptyline, but not of Fluoxetine, into Brain is enhanced in Mice with Blood-Brain Barrier Deficiency due to mdr1a P-Glycoprotein Gene Disruption. Neuropsychopharmacology. 2000; 22: 380-387.
- Uhr M, Tontsch A, Namendorf C, Ripke S, Lucae S, Ising M, et al. Polymorphisms in the Drug Transporter Gene ABCB1 predict Antidepressant Treatment Response in Depression. Neuron. 2008; 57: 203-209.
- Breitenstein B, Brückl T, Ising M, Müller-Myhsok B, Holsboer F, Czamara D. ABCB1 Gene Variants and Antidepressant Treatment Outcome: A Meta- Analysis. AmJMedGenetB Neuropsychiatr Genet. 2015; 168B: 274-283.
- Breckenridge A, Aronson J, Blaschke T, Hartman D, Peck C, Vrijens B. Poor Medication Adherence in Clinical Trials: Consequences and Solutions. Nat Rev Drug Discov. 2017; 16: 149-150.
- Schatzberg A, DeBattista C, Lazzeroni L, Etkin A, Murph G, Williams L. ABCB1 Genetic Effects on Antidepressant Outcomes: a Report from the iSPOT-D Trial. Am J Psychiatry. 2015; 172: 751-759.
- Dong C, Wong ML, Licinio J. Sequence variations of ABCB1, SLC6A2, SLC6A3, SLC6A4, CREB1, CRHR1 and NTRK2: Association with Major Depression and Antidepressant Response in Mexican-Americans. Mol Psychiatry. 2009; 14: 1105-1118.
- Breitenstein B, Scheuer S, Brückl T, Meyer J, Ising M, Uhr M, et al. Association of ABCB1 Gene Variants, Plasma Antidepressant Concentration, and Treatment Response: Results from a Randomized Clinical Study. J Psychiatr Res. 2016; 73: 86-95.
- Perlis RH, Fijal B, Charia S, Heinloth AN, Houston JP. Failure to Replicate Genetic Associations with Antidepressant Treatment Response in Duloxetinetreated Patients. Biol Psychiatry. 2010; 67: 1110-1113.
- Breitenstein B, Scheuer S, Pfister H, Uhr M, Lucae S, Holsboer F, et al. The Clinical application of ABCB1 Genotyping in Antidepressant Treatment: A Pilot Study. CNS Spectr. 2014; 19: 165-175.
- Holsboer-Trachsler E, Hättenschwiler J, Beck J, Brand S, Hemmeter U, Keck M, et al. Die Akutbehandlung depressiver Episoden. Swiss Medical Forum. 2016; 16: 716-724.
- Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmöller J, Johne A, et al. Functional Polymorphisms of the Human Multidrug-Resistance Gene: Multiple Sequence Variations and Correlation of one Allele with P-Glycoprotein Expression and Activity in vivo. Proc Natl Acad Sci USA. 2000; 97: 3473-3478.
- Winner JG, Carhart JM, Altar CA, Allen JD, Dechairo BM. A Prospective, Randomized, Double-Blind Study assessing the Clinical Impact of Integrated Pharmacogenomic Testing for Major Depressive Disorder. 2013; 16: 219- 227.