
Research Article
J Dermatolog. 2014;1(6): 1031.
Langerhans Cell Histiocytosis and Myelodysplastic Syndrome: A Casual Association or a Pathogenetic Correlation?
Resende C1*, Marques H2, Pereira T1, Pardal F3, Torres F4 and Paiva A5
1Dermatology and Venereology Department, Hospital de Braga, Portugal
2Oncology Department, Hospital de Braga, Portugal
3PathologyDepartment, Hospital de Braga, Portugal
4CGC Genetics, Portugal
5Institute for Blood and Transplantation, Portugal
*Corresponding author: Resende C, Department of Dermatology and Venereology, Hospital de Braga, Braga, Sete Fontes – So Victor. 4710-243 Braga, Portugal
Received: November 10,2014; Accepted: December 01,2014; Published: December 03, 2014
Abstract
Background: Langerhans Cell Histiocytosis (LCH) is a rare disease in adults, characterized by a proliferation of abnormal Bone Marrow (BM) derived Langerhans Cells (LCs) in single or multiple organs. The Myelodysplastic Syndrome (MDS) represents a group of clonal disorders, characterized by dysplasia in two or more lineages. We try to clarify if LCH and MDS have the same clonal origin or if they are two independent neoplasms.
Methods: We report a case of a female, with a4-month history of pruriticred to brownish papules, localized on trunk and proximal limbs, covered by crusts. She was diagnosed with LCH, with involvement of skin, lymph nodes, lungs and liver. Two months later, by studying a progressive pancytopeniathe diagnosis of a high-risk MDS was done. We did a molecular examination of BM to detect the presence of BRAF (V600E) mutation and a flow cytometrycell sorting to detect the presence of mutation in plasmacytoid dendritic cells, myeloid dendritic cells and CD34+ cells.
Results: The BRAF mutation (V600E) was detected in BM cells as well asinpurified plasmacytoid dendritic cells and it wasn´t detected in purified myeloid dendritic cells. The patient died 8 months later with a bacterial infection.
Conclusion: We found no evidence of clonal relationship between LCH and MDS. These results don´t support the hypothesis of a common stem cell origin for the two neoplasms.
Keywords: Langerhans Cell Histiocytosis; Myelodysplastic syndrome; BRAF mutation; Bone marrow
Abbreviations
LCH: Langerhans Cell Histiocytosis; BM: Bone Marrow; LCs: Langerhans Cells; MDS: Myelodysplastic Syndrome; CT: Computed Tomography; PET: Positron Emission Tomography; PCR: Polymerase Chain Reaction; WHO: World Health Organization
Introduction
Background
Langerhans Cell Histiocytosis (LCH) is a rare disease of unknown etiology, affecting patients of all ages, with an estimated incidence of approximately 2-5 per million per year in infants and children and is even rarer in adults [1-3], but more definitive epidemiological data are needed, since many cases of localized LCH are likely undiagnosed [1].
LCH belongs to a spectrum of histiocytic disorders, which have in common the involvement of the mononuclear-phagocyte system and a proliferation of abnormal, Bone Marrow (BM) derived Langerhans Cells (LCs) in single or multiple organs [1,4,5]. Whether the infiltrating cells are truly neoplastic or reactive in nature is still a matter of debate [3]. Badalian-Very and colleagues demonstrate that 57% of LCH specimens display mutations in BRAF, which encode a known oncogenic V600E form of the BRAF gene. The detection of clonal histiocytes in different clinical forms of LCH seems to indicate a neoplastic disorder arising from mutations in BM precursor cells [1].
The Myelodysplastic Syndrome (MDS) represents a heterogeneous group of clonal disorders, characterized by dysplasia in 2 or more lineages and increased risk of acute leukemia transformation [6], all of which have their origin in multipotential hematopoietic stem cells. Patients usually present with peripheral blood cytopenias and hyper cellular BM, although hypo cellular BM is present in one quarter of the cases [6].
We report a case of a patient with a LCH, with involvement of skin, lymph nodes, lungs and liver. Two months after the diagnosis of LCH, the patient developed a severe pancytopenia, caused by highrisk MDS, with dysplasia of erythroid and granulocytic lineages.
In this case, we tried to answer the question if the LCH and MDS have origin in same stem cell or if they have origin in two different stem cells, and consequently they are 2 independent hematologic neoplasms.
Material and Methods
A previously healthy, 71-year-old Caucasian woman, presented with a 4-month history of pruritic, non-tender papules, with a color variation from red to brownish, localized on her trunk, abdomen and her proximal limbs, covered by crusts (Figure 1a, Figure 1b). The lesions were isolated and some were confluent and had an uneven surface, with a rough texture. She had no nail, hair, teeth, or mucosal alterations. The remainder of the examination was also normal.
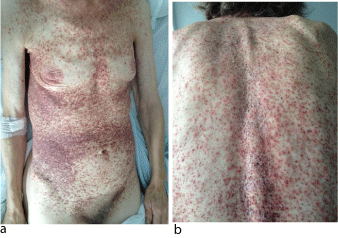
Figure 1: Lesions in a 71-year-old woman with LCH. (a,b) . Papules, with a
color variation from red to brownish, localized on trunk, covered by crusts.
She was not taking any medication. Her past medical history was unremarkable and there was no family history of cutaneous or hematologic diseases. Histopathologic examination of an incisional biopsy of a lesion taken from the anterior part of the trunk revealed large ovoid cells contain abundant eosinophilic cytoplasm and reniform nucleus, suggestive of LCH (Figure 2a, Figure 2b).
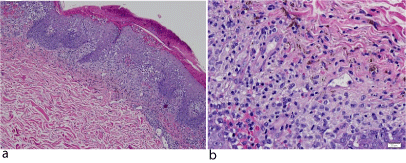
Figure 2: Histologic analysis of a lesion taken from the anterior part of the
trunk, revealing large ovoid cells, containing abundant eosinophiliccytoplasm
and reniform nucleus, suggestive of LCH. Hematoxylin and eosin stain;
magnification (a) x100 and (b) (x400).
Clinical staging procedures including complete hematologic examination with BM biopsy, chest, abdominal, pelvic Computed Tomography (CT), cerebral magnetic resonance and whole body Positron Emission Tomography (PET) scan. The examinations showed mediastinicadenopathies, with 3,5 cm of diameter and a subcarinaladenopathy with 4 cm of diameter and hepatosplenomegaly. The PET scan showed hypercaptation of fludeoxy glucose (18F) in supra and infra-diafragmatic lymph nodes, liver and lungs.
Taken together, clinical, pathological and imagiological examinations, suggested the diagnosis of LCH, with involvement of skin, lymph nodes, lungs and liver.
The molecular examination of BM by multiplex Polymerase Chain Reaction (PCR) detected the V600E mutation in BRAF gene. Genomic DNA was extracted from cell populations using the Citogene® DNA isolation kit (Citomed, Portugal). PCR amplification of BRAF exon 15 was performed in a Veriti® 96-Well Thermal Cycler (Applied Biosystems, Foster City, CA, USA) as follows: each reaction contained approximately 10ng of template DNA, 22.5uL of Platinum® PCR Super Mix (InvitrogenTM Life Technologies, Carlsbad, CA, USA) and 5pmol of each primer (BRAF-15 Forward: 5’–ACTCTTCATAATGCTTGCTC–3’ and BRAF-15 Reverse: 5’ – CTTTCTAGTAACTCAGCAGC – 3’) in a final volume of 25uL. PCR cycling parameters were 1 cycle of 94 °C for 10 minutes, followed by 35 cycles of 94 °C for 15 seconds, 55 °C for 30 seconds, and 72 °C for 60 seconds and, finally, 1 cycle of 72 °C for 10 minutes. When needed, a nested PCR was done, using the same reagents and conditions except for the template DNA (1-2uL of PCR product instead of genomic DNA). PCR products were analyzed in a QIAxcel advanced apparatus (QIAGEN, Hilden, Germany).After visual confirmation of amplification, 2uL of the PCR product were purified with 1uL of illustra TMExoProStarTM 1-Step (GE Healthcare, Amersham Place, Buckinghamshire, UK) and analyzed by bidirectional sequencing on an 3500xL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), using the ABIPRISM® BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Mutational analysis was done using SeqScape® Software v2.7 (Applied Biosystems, Foster City, CA, USA).
Her initial blood cell counts were as follows: white blood cells 5.1×109/L; hemoglobin 11.8 g/dL and platelets 41×109/L.
The patient received an initial treatment of continuous oral prednisone (40 mg/m2 daily in 3 doses for 4 weeks, tapering over 2 weeks) and vinblastine (6 mg/m2 intravenous bolus, weekly for 6 weeks). After two months of therapy, the cutaneous lesions aggravated. However, at this time, the laboratory findings showed a pancytopenia (white blood count 1,2x109 /L), a normocytic and normochromic anemia (hemoglobin 6,6 g/dL), a thrombocytopenia (platelet count <10x109/L) and consequently, the vinblastine was stopped.
At this time, 6 months after the diagnosis of LCH, the BM aspirate was normocellular, with 24% of erythroblasts, 61% of neutrophils, 5% of monocytes, 3% of lymphocytes, 1% of eosinophils and 5,6% of blasts cells. Immunophenotypic analysis of BM as pirate suggested dysplasia in granulocytic and erythroid lineages, based on an aberrant expression pattern of CD13/CD11b/CD16 and CD105/CD71/CD36 respectively.
The BM biopsy showed a hyper cellular BM, with almost total absence of adipocytes, with trilineage hematopoiesis. The myeloid lineage showed increase of precursor’s cells, but diminution of maturation (Figure 3a). In the erythroid lineage, there was an increase of erythroidprecursors. We also observed large ovoid cells containing abundant eosinophiliccytoplasm and reniform nucleus. Occasional binucleate forms were present (Figure 3b). Theseinfiltrates were intimately admixed with the hematopoietic tissue. Immunostainings were positive forCD1a and S100 protein (Figure 3c, Figure 3d), indicated a Langerhans cell-phenotype and suggesting invasion of the BM by histiocytes. The cytogenic BM analysis didn´t show any chromosomal abnormalities.
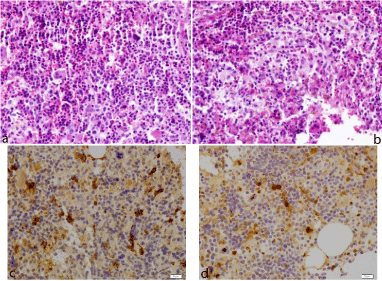
Figure 3: Histologic analysis of the BM showed a hyper cellular BM, with
trilineage hematopoiesis and the myeloid lineage showed increase of
precursor’s cells, but diminution of maturation. Hematoxylin and eosin stain;
magnification (x400) (Figure 3a).
Histologic analysis of the BM alsorevealed large ovoid cells contain abundant
eosinophiliccytoplasm and reniform nucleus, suggestive of LCH..Hematoxylin
and eosin stain; magnification (x400) (Figure 3b).
Immunochemical staining were positive for S100 protein (Figure 3c) and
CD1a (Figure 3d), indicated a Langerhans cell-phenotype and suggesting
invasion of the BM by histiocytes. Magnification (c,d) (x400).
These findings supported the diagnosis of LCH and of a refractory anemia with excess of blasts-1 by the World Health Organization (WHO) classification, with an intermediate risk (2,0) of International Prognostic Scoring System.
With the objective of understand if LCH and MDS have the same origin or if they are 2 independent hematologic neoplasms, a flow cytometrycell sorting was performed followed by an amplification of Deoxyribonucleic Acid (DNA) by PCR and gene sequencing of BRAF region, where is located V600Emutation, in purified plasmacytoid dendritic cells, myeloid dendritic cells and CD34+ cells. Figure 4 showed the flow cytometry of the three studied populations for the presence of BRAF mutation. This mutation was only observed in purified plasmacytoid dendritic cells and not in myeloid dendritic cells. It was not possible to study the presence of the mutation in purified CD34+ cells, due to low amounts of DNA.
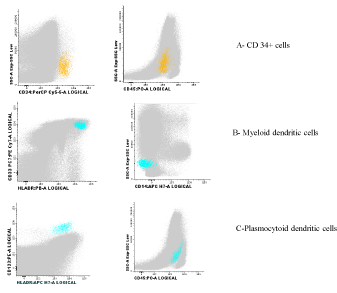
Figure 4: Flow cytometry of cells studying, where the BRAF mutation was studied: A- CD34+ cells, B-Myeloid dendritic cells, C- Plasmocytoid dendritic cells.
Treatment with azacitidine had been initiated (75 mg/m2s.c. for 7 consecutive days monthly). The patient did not achieve a hematologic response to treatment and her clinical condition deteriorated. She died 8 months later with pancytopenia and severe bacterial infection.
Discussion
Histiocytic disorders are a group of rare diseases that involve histiocytes, which are derived from BM stem cells [7]. The Langerhans cells are an important antigen presenting cell and differ from other histiocytes in being CD1a-positive [7]. The etiology of LCH remains unknown.
It was consider that LCH results from immune dysfunction, since several cytokines are present at higher levels in the lesions than in normal surrounding tissue [1,7]. More recently, the detection of clonal histiocytes in different clinical forms of LCH seems to indicate a neoplastic disorder arising from mutations of BM precursor cells [1]. Badalian-Very and colleagues demonstrate that 57% of LCH specimens display mutations in BRAF, which encode a known oncogenic V600E form of the BRAF. This study also demonstrated the specificity of BRAF V600 E for pathologic LCs and it is not present in normal proliferating LCs [8]. Our patient had the V600E mutation in BRAF gene, detected in blood and marrow specimens, which could contribute to the developing of LCH.
The occurrence of LCH in the elderly population is rare and involvement limited to skin in this age group is even more unusual [9].The diagnose of our patient was obtained by the dermatologic physical examination and cutaneous biopsy. Similarly to large studies, the most common skin lesions were papules with brown, red or crusted areas on the trunk [3,9,10].
The clinical picture of LCH is heterogeneous, ranging from a single system involvement, to a multisystem life-threating disease, with proliferation of abnormal and clonal LCs in skin, bone, lymph nodes, lungs, liver, spleen and BM [3,4]. Clinical staging of our patient revealed also involvement of lymph nodes, lungs and liver.
The prognosis of LCH is often unpredictable [3,5]. Patients with disease that is localized to one organ system have a good prognosis and seem to need minimal or even no treatment [5]. In contrast, multiple organ involvement carries a risk of a poor outcome, including 10-20% mortality and a 50% risk of life-impairing morbidity [5].
The effective treatment for LCH remains controversial at present [1]. In the literature, vinblastine, vincristine, prednisolone and cyclophosphamide, either alone, or in combinations, are mentioned in the treatment of disseminated disease [1,11], but the standard of care is the combination of vinblastine and prednisolone [12]. Nevertheless, no modality has clearly shown convincing results, with remission rates varying between 21 to 65% [1]. Targeting the BRAF oncogene provides a new therapeutic opportunity for BRAFmutated tumors and the clinical efficacy of BRAF kinase inhibitors has already been reported in advanced melanoma [13]. There are also reports of efficacy of vemurafenib in anecdotal cases of patients with LCH. The toxicity profile of the current generation of BRAF V600 E inhibitors includes skin cancers, which are likely to be mechanismbased effects of the drugs [12]. Clinical trials will have to be carefully planned to optimize the risk benefit ratio of BRAF V600E inhibition in this population [12].Our patient received an initial treatment of oral prednisone and intravenous vinblastine, without good results.
Patients with LCH have a higher than normal risk of developing secondary cancers [7]. Concurrent LCH/second malignancy has been reported in few patients, and some patients have had their second malignancy first, and developed LCH at a later date [7]. Solid tumors associated with LCH include retinoblastoma, brain tumors, hepatocellular carcinoma, and Ewing´s sarcoma [7]. There is also an association between LCH and MDS/myeloblastic leukemia [11]. They can either co-exist in the same patient or one of them may precede the other [11].
Many theories have been issued to explain this association [7]. A genetic predisposition can also be a possible explanation. Secondary MDS/ acute leukemia could develop after treatment of LCH by chemotherapy (etoposide, vinblastine) [7]. The cumulative risk of MDS increases by 0,25%-2% per year from 1-2 years after start of therapy and continues up to 6-8 years after its cessation [15,16]. Consequently, in our case, the hypothesis of a MDS secondary to vinblastine is improbable, since the latency period is only two months.
An interesting association of MDS/acute leukemia and LCH could be the anomalous cytokine production and a reactive histiocyte reaction [3].
Recent studies on global gene expression patterns in LCH support the notion that the disease may arise from early myeloid precursors rather than mature LCs themselves [12-14], and consequently we could expect that the BRAF mutation (V600E) would be found in purified myeloid dendritic cells, which could explain, at least in part, the association between LCH and MDS. In this particular the study of BRAF mutation (V600E) was only observed in purified plasmacytoid dendritic cells, which didn´t support our hypothesis. It was not possible to study the presence of mutation in purified CD34+ cells, because the obtained DNA amount was not sufficient to do this analysis. However a mutation on the early common precursor must be present in both neoplasic populations [12,15,16]. Proliferation of plasmacytoid dendritic cells in lymph nodes or extra nodal sites, associated with myeloid neoplasms, including myelodisplastic syndrome has been described in the literature [17]. LCs, another type of dendritic cell, also can proliferate in myeloproliferative disorders. It was hypothized by Song HL and colleagues that the combined proliferations of plasmacytoid dendritic cells and LCs could derive from the same hematopoietic stem cells but they could differentiate divergently under the effect of different micro environments [17].
To the best of our knowledge this is the first study that tries to clarify if the LCH and MDS have the same origin or if they are 2 independent hematologic neoplasms, studying the BRAF mutation, after cell purification by cell sorting.
The risk of MDS increases with advancing age; approximately 86% of patients with newly diagnosed MDS in the United States are > 60 years old [18]. Consequently, another hypothesis is that our old patient and most of the cases described in medical literature the association of the two independent diseases happen by chance.
The clinical manifestations of MDS are highly variables, depending on the MDS subtype and it ranges from indolent to life threatening [18]. Anemia (typically macrocytic) is the most common peripheral blood abnormality, occurring in approximately 80% to 85% of patients. Thrombocytopenia occurs in around 30% to 45% of MDS cases, with approximately 40% of patients found to have neutropenia at diagnosis. The first sign of MDS in our patient was a severe pancytopenia.
A combination of morphology to detect multilineage dysplasia in the BM and peripheral blood and cytogenetic, to detect characteristic clonal abnormalities, are used in establishing a diagnosis of MDS [15,19]. In our patient, these findings support the diagnosis of a refractory anemia with excess of blasts-1 by the World Health Organization (WHO) classification.
Most patients with MDS were treated historically with supportive measures only [15]. The approval of agents for treatment of MDS includes the DNA methyl transferase inhibitors azacitidine and decitabine [15], as well as the immunomodulatory agent lenalidomide [15]. The use of allogeneic hematopoietic stem cell transplantation remains the only curative modality for patients with MDS but it is only available to young patients [15]. Unfortunately, the majority of patients with MDS are not candidates for transplantation, due to age, comorbidities and lack of suitable donors [15]. Our patient received treatment with azacitidine. A randomized controlled trial compares azacitidine, with supportive or conventional care in patients with high risk MDS, results in significant improvements in response rates, overall survival and quality of life [20].
Mortality due to infection is the main cause of death of MDS [16]. Our patient died with neutropenic fever and septicemia.
This case of almost simultaneous occurrence of LCH and MDSillustrates a rare association and allowed us to study different clonal and non-clonal BM population to evaluate the possible cellular and molecular causality between them. In this case, we can´t neither affirm, nor exclude an evidence of clonal relation between LCH and MDS and it´s necessary more studies in future, analyzing the relationship between LCs and plasmacytoid dendritic cells.
References
- Saiz A, Martinez MA, Grande C, Vanaclocha F. Langerhans' cell histiocytosis in an adult with acute myelogenous leukaemia. Virchows Arch. 2004; 445: 93-95.
- Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010; 116: 1919-1923.
- Stefanato CM, Andersen WK, Calonje E, Swain FA, Borghi S, Massone L, et al. Langerhans cell histiocytosis in the elderly: a report of three cases. J Am Acad Dermatol. 1998; 39: 375-378.
- Arico M. Langerhans cell histiocytosis in adults: more questions than answers? Eur J Cancer. 2004; 40: 1467-1473.
- Abla O, Egeler RM, Weitzman S. Langerhans cell histiocytosis: Current concepts and treatments. Cancer Treat Rev. 2010; 36: 354-359.
- Stetler-Stevenson M, Arthur DC, Jabbour N, Xie XY, Molldrem J, Barrett AJ, et al. Diagnostic utility of flow cytometric immunophenotyping in myelodysplastic syndrome. Blood. 2001; 98: 979-987.
- Ghosn MG, Haddad AC, Nassar MN, Abadjian GA, El Karak FR, Aftimos PG. Acute myeloid leukemia and Langerhans' cell histiocytosis: multiple theories for an unusual presentation. Leuk Res. 2010; 34: 406-408.
- Badalian-Very G, Vergilio JA, Fleming M, Rollins BJ. Pathogenesis of Langerhans cell histiocytosis. Annu Rev Pathol. 2013; 8: 1-20.
- Surico G, Muggeo P, Rigillo N, Gadner H. Concurrent Langerhans cell histiocytosis and myelodysplasia in children. Med Pediatr Oncol. 2000; 35: 421-425.
- Wollina U, Kaatz M, Kronert C, Schonlebe J, Schmalenberg H, Schreiber G, et al. Cutaneous Langerhans cell histiocytosis with subsequent development of haematological malignancies. Report of two cases. Acta Dermatovenerol Alp Pannonica Adriat. 2006; 15: 79-84.
- Ventura F, Pereira T, da Luz Duarte M, Marques H, Pardal F, Brito C. Indeterminate cell histiocytosis in association with acute myeloid leukemia. Dermatol Res Pract. 2010; 569345.
- Badalian-Very G, Vergilio JA, Degar BA, Rodriguez-Galindo C, Rollins BJ. Recent advances in the understanding of Langerhans cell histiocytosis. Br J Haematol. 2012; 156: 163-172.
- Tadmor T, Tiacci E, Falini B, Polliack A. The BRAF-V600E mutation in hematological malignancies: a new player in hairy cell leukemia and Langerhans cell histiocytosis. Leuk Lymphoma. 2012; 53: 2339-2340.
- Egeler RM, Neglia JP, Arico M, Favara BE, Heitger A, Nesbit ME, et al. The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other solid tumors. The LCH-Malignancy Study Group of the Histiocyte Society. Hematol Oncol Clin North Am. 1998; 12: 369-378.
- Zeidan AM, Faltas B, Douglas Smith B, Gore S. Myelodysplastic syndromes: what do hospitalists need to know? J Hosp Med. 2013; 8: 351-357.
- Romeo M, Chauffaille MLF, Silva MRR, Kerbauy J. Myelodysplastic syndromes: histopathology as prognostic factor.Rev. Bras. Hematol. Hemoter. 2001; 23: 63-68.
- Song HL, Huang WY, Chen YP, Chang KC. Tumorous proliferations of plasmacytoid dendritic cells and Langerhans cells associated with acute myeloid leukaemia. Histopathology. 2012; 61: 974-983.
- Foran JM, Shammo JM. Clinical presentation, diagnosis, and prognosis of myelodysplastic syndromes. Am J Med. 2012; 125: 6-13.
- Orazi A. Histopathology in the diagnosis and classification of acute myeloid leukemia, myelodysplastic syndromes, and myelodysplastic/myeloproliferative diseases. Pathobiology. 2007; 74: 97-114.
- Kim YJ, Jang JH, Kwak JY, Lee JH, Kim HJ. Use of azacitidine for myelodysplastic syndromes: controversial issues and practical recommendations. Blood Res. 2013; 48: 87-98.