
Case Report
Austin Endocrinol Diabetes Case Rep. 2016; 1(1): 1002.
A Case of IgG4-Related Infundibulo-Hypophysitis Presented as Pituitary Incidentaloma
Wat WZM¹*, Cheng Y² and Kho BCS¹
¹Department of Medicine, Pamela Youde Nethersole Eastern Hospital, Hong Kong
²Department of Clinical Pathology, Pamela Youde Nethersole Eastern Hospital, Hong Kong
*Corresponding author: WAT Zee Man, Department of Medicine, Pamela Youde Nethersole Eastern Hospital, 3 Lok Man Road, Chai Wan, Hong Kong
Received: May 25, 2016; Accepted: July 13, 2016; Published: July 15, 2016
Abstract
IgG4-related infundibulo-hypophysitis is very rare and is usually presented with hypopituitarism, central diabetes insipidus or pseudotumour with mass effect. We reported a case presented as a large pituitary incidentaloma with height of 17mm in the MRI orbits of a 45 year-old male complaining of bilateral chronic upper eyelid swelling and generalised lymphadenopathy. His upper eye lid biopsy showed reactive lymphocytic infiltrates and lymph node biopsy reactive follicular hyperplasia. The pituitary mass, lymphadenopathy and eye signs dramatically responded to a therapeutic trial of prednisolone. His serum IgG4 level was later found significantly elevated and retrospective IgG and IgG4 immunohistochemistry study in previous cervical lymph node and nasopharyngeal biopsies confirmed the diagnosis of IgG4-related disease. His pituitary incidentaloma is likely IgG4-related infundibulo-hypophysitis. Repeat MRI after two months’ of steroid treatment showed the pituitary lesion reduced to a 7mm stalk nodule. Serial MRI showed that the stalk nodule remained static in size with low dose maintenance steroid.
Keywords: Pituitary mass; IgG4 related disease; Infundibulo-hypophysitis; Hypophysitis
Abbreviations
CT: Computed Tomography; MRI: Magnetic Resonance Imaging; IgG4: Immunoglobulin G4; NR: Normal Range; ESR: Erythrocyte Sedimentation Rate; CRP: C-Reactive Protein; ANA: Anti-Nuclear Antibodies; C3: Complement 3; LH: Luteinizing Hormone; FSH: Follicle Stimulating Hormone; TSH: Thyroid-Stimulating Hormone; HE: Haematoxylin Eosin
Case Presentation
The patient was first presented in April 1999, at the age of 45 years, with 2 years’ history of cervical lymphadenopathy and bilateral swollen upper eyelids. Biopsy of the cervical lymph node in March 2000 reported reactive follicular hyperplasia. Blood tests revealed only mild lymphocytosis of 4.81x 109/L (NR 0.6-3.4) and mild eosinophilia 0.84 x109/L (NR 0-0.7). Other blood tests including ESR, CRP, ANA pattern, C3, Rheumatoid factors were all normal. His symptoms did not respond to antibiotics.
He later defaulted follow up and returned in 2003 due to persistent symptoms. While the CT orbits in May 2003 showed bilateral prominent lacrimal glands and swollen extra-ocular muscles, his left lacrimal gland biopsy in January 2005 was unremarkable. However, his follow up MRI (1.5 Tesla) orbits in November 2005 incidentally revealed a large homogenously contrast enhanced pituitary mass of 17mm in height, moderately impressing on the optic chiasm (Figure1). His visual field by confrontation was full. He did not have any symptoms suggestive of hypopituitarism or diabetes insidipus. His blood tests in December 2005 showed mild hypogonadotrophic hypogonadism with serum total testosterone level of 5.4 nmol/L (NR 10-35), LH 3.9 IU/L (NR 1-12) and FSH 4.8 IU/L (NR 1-12). Other hormonal axes were normal with serum level of prolactin 344mU/L (NR 38-555), 10am cortisol 242nmol/L (NR 123-645), thyroxine 11.5pmol/L (NR 9.1-23.8) and TSH 1.44mIU/L (NR 0.32-5). Inpatient monitoring of the urine output and osmolarity revealed no diabetes insipidus. His immunoglobulin pattern showed significantly raised serum IgG level of 35.6g/L (NR 6.94-16.18g/L), but serum electrophoresis revealed no monoclonal band and urine no Bence Jones protein.
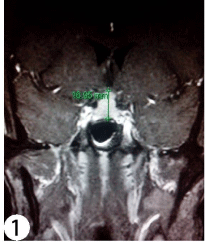
Figure 1: Post-contrast T1-weighted MRI orbits of the patient showing
an enlarged contrast enhancing pituitary gland of around 17mm height
impressing on the optic chiasm. The normal infundibulum could not be
identified.
Multiple tissue biopsies were subsequently performed. Fine needle aspiration of the left submandibular lymph node and nasopharyngeal biopsy were done in November 2005 as his MRI also revealed lymphadenopathy up to 2cm in size in bilateral submandibular, cervical and retropharyngeal regions, and thickening of the posteriorlateral walls of the nasopharynx. Both specimens revealed findings compatible with reactive lymphoid hyperplasia (Figure 2). Excisional biopsy of right cervical lymph node in December 2005 similarly displayed features of reactive follicular hyperplasia with peri-nodal fibrosis and adhesion to submandibular gland. Increased number of plasma cells was observed in the inter-follicular zone, but no light chain restriction was detected. His right upper eyelid biopsy in January 2006 showed findings suggestive of autoimmune dacryoadenitis with lobules of lacrimal glands containing dense lymphoplasmacytic infiltrate, atrophy of acini and mild interlobular fibrosis. Occasional foci of lymphoepithelial lesions and a few reactive lymphoid follicles were observed. His contrast CT thorax and abdomen in January 2006 showed shotty mediastinal, bilateral hilar and para-aortic lymph nodes up to 1.6cm in size
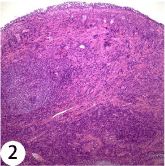
Figure 2: HE stain showing lymphoplasmacytic infiltration in the nasopharynx
specimen.
A therapeutic trial of prednisolone 30mg daily (~0.5mg/kg/day) was subsequently given in January 2006 and dramatic improvement of eyelid swelling was observed just after few days of treatment. The MRI pituitary gland in March 2006, after two months’ of steroid treatment, also showed drastic shrinkage of the sellar lesion to an infundibular mass of size 8mm x 7mm. The pituitary gland was no longer enlarged. Prednisolone was later gradually tailed down to a low maintenance dose of 5mg daily. However, in June 2006, the patient stopped the steroid himself and eye symptoms soon relapsed 2 weeks later but rapidly resolved again after medication resumed. The MRI orbits in August 2006 confirmed marked reduction in the hypertrophy of all extra-ocular muscles and swelling of both lacrimal glands. The MRI pituitary gland in August 2007 showed the infundibulum nodule remained static in size. Testosterone replacement was later started in October 2007 in view of his serum total testosterone level in May 2007 remained low at <0.03nmol/L in spite of resolution of the pituitary mass and normal serum prolactin level of 397mU/L.
The patient’s serum IgG4 level was later found elevated at 400mg/ dl (NR 0-291mg/dl) in November 2009 when blood test for IgG4 serology was accessible. Immunohistochemical study of his previous cervical lymph node and nasopharyngeal specimens revealed both increased absolute number of IgG4 bearing plasma cells (>50/high power field) and increased IgG4/IgG ratio of 90% (Figure 3a & 3b). The diagnosis of IgG4-related disease was then confirmed. His pituitary incidentaloma is probably IgG4-related infundibulohypophysitis. Long term maintenance dose of prednisolone 5mg daily was continued and the disease activity remained well controlled clinically and radiologically. Formal perimetry done in February 2010 revealed no visual field defect. His CT abdomen and thorax in 2011 detected no more lymphadenopathy. His latest MRI of pituitary gland in January 2015 showed the pituitary stalk nodule remained static in size (Figure 4).
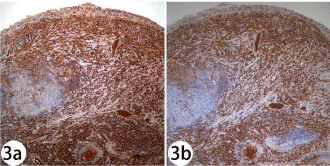
Figure 3: 3a & 3b: Immunohistochemical staining for IgG (Figure 3a) and
IgG4 (Figure 3b) showing the number of IgG4 positive plasma cells in the
nasopharynx increased and the ratio of IgG4 to IgG plasma cells being about
90%. With compatible morphology, absolute number of IgG4 positive plasma
cells >50 per high power field and IgG4/IgG ratio >40% are essential for
histological diagnosis [16].
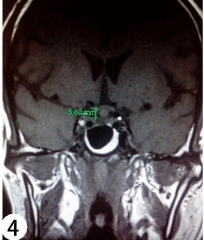
Figure 4: Non-contrast T1-weighted MRI pituitary gland of the patient
showing an isointense pituitary stalk nodule of size around 6mm touching the
optic chiasm. The pituitary gland was not enlarged.
Discussion
IgG4-related hypophysitis is very rare and was first described in 2004. Up till now, thirty-eight reported cases were found in the literature [1-9]. All of them presented with pituitary pseudotumour with mass effect, hypopituitarism or central diabetes insipidus. Among these 38 cases, hypopituitarism was observed in 32 cases (84.2%) and diabetes insipidus in 26 cases (68.4%). Twenty-two cases (57.9%) had both hypopituitarism and diabetes insipidus. We hereby reported the first case of IgG4 related hypophysitis presented as pituitary incidentaloma. Our patient was asymptomatic and only mild biochemical hypogonadotrophic hypogonadism with serum testosterone level of 5.4nmol/ L (NR 10-35) was detected on presentation.
IgG4-related disease was first described in 2001 by Hamno et al in a patient with sclerosing pancreatitis [10]. It is a fibro-inflammatory tumefactive condition with presence of dense lymphoplasmacytic infiltration, rich IgG4-positive plasma cells and stori form fibrosis. Plasma IgG4 concentration is often, though not always, raised [11]. The exact etiology of the disease remains unclear, though allergic reaction and autoimmune mechanisms have been proposed.
Multi-organ involvement is common in IgG4-related disease, though single organ manifestation is possible. Lacrimal glands and orbital soft tissue are commonly affected sites as illustrated in our case [12,13]. It is known that pituitary gland and stalk are very rarely involved in IgG4-related diseases. In 2011, Leporati et al proposed a set of diagnostic criteria for IgG4-related hypophysitis [14]. They include 1) the presence of mononuclear cell infiltrates within the pituitary gland rich in plasma cells and lymphocytes, with more than 10 IgG4 positive cells; or 2) a sellar mass and/ or thickened pituitary stalk with biopsy proven IgG-4 related diseases involving other organs; or 3) raised serum IgG4 level (> 140mg/dL), and the sellar and/ or stalk mass reduced in size and symptoms improved with corticosteroid treatment. Our case was diagnosed based on the second and third criteria.
Recent data suggested the prevalence of IgG4-related hypophysitis might be underestimated. In a Japanese study, it was diagnosed in 4% of 170 patients with hypopituitarism and/or central diabetes insipidus screened and in 30% (7 out of 23 patients) of hypophysitis cases [9]. Clinically, IgG4-related disease predominately occurs in middle aged and elderly male [2] while lymphocytic hypophysitis typically affects females at the age of 30 to 40 years, usually during late pregnancy or early postpartum period [15]. Isolated IgG4-related hypophysitis can be differentiated from lymphocytic hypophysitis by histology as the latter though also reveals extensive lymphocytic infiltration, plasma cells are only the minor population in contrast to IgG4-related diseases. Immunostaining with IgG and IgG4 antibodies can always confirm the diagnosis of IgG4-related hypophysitis.
Correct diagnosis of IgG4-related hypophysitis has important therapeutic and prognostic implications. In the context of a contrast enhancing pituitary and/or stalk lesion with raised serum IgG and IgG4 level, a therapeutic trial of oral steroid can be considered before proceeding to invasive biopsy. As systemic involvement is common in IgG4-related disease, searching for more easily accessible sites of involvement such as lymph nodes for biopsy is also possible to avoid pituitary biopsy as illustrated in our case.
Although IgG4-related infundibulo-hypophysitis is very sensitive to corticosteroid therapy, steroid withdrawal usually accompanies relapse as demonstrated in our case. Hence, drug compliance should be emphasized in patient care as long term low dose steroid is needed for disease control.
References
- Iseda I, Hida K, Tone A, Tenta M, Shibata Y, Matsuo K, et al. Prednisolone markedly reduced serum IgG4 levels along with the improvement of pituitary mass and anterior pituitary function in a patient with IgG4-related infundibulohypophysitis. Endocrine Journal. 2014; 61: 195-203.
- Ohkubo Y, Sekido T, Takeshige K, Ishi H, Takei M, Nishio S, et al. Occurrence of IgG4-related hypophysitis lacking IgG4-bearing plasma cell infiltration during steroid therapy. Internal Medicine. 2014; 53: 753-757.
- Imber BS, Lee HS, Kunwar S, Blevins LS, Aghi MK, et al. Hypophysitis: a single-centre case series. Pituitary. 2015; 18: 630-641.
- Bando H, Iguchi G, Fukuoka H et al. A diagnostic pitfall in IgG4-related hypophysitis: infiltration of IgG4-positive cells in the pituitary of granulomatosis with polyangiitis. Pituitary. 2015; 18: 722-730.
- Sosa GA, Bell S, Christiansen SB, Pietrani M, Glerean M, Loto M, et al. Histologically confirmed isolated IgG4-related hypophysitis: two case reports in young women. Endocrinol. Diabetes and Metab Case Rep. 2014.
- Harano Y, Honda K, Akiyama Y et al. A case of IgG4-related hypophysitis presented with hypopituitarism and diabetes insipidus. Clinical medicine Insights: Case reports. 2015; 8: 23-26.
- Batista RL, Ramos LS, Cescato VA, Musolino NR, Borba CG, Silva GO, et al. Thickened pituitary stalk associated with a mass in the sphenoidal sinus: an alarm to suspect hypophysitis by Immunoglobulin G4? Int Arch Otorhinolargngol. 2015; 19: 273-276.
- Ngaosuwan K, Trongwongsa T, Shuangshoti S. Clinical course of IgG4- related hypophysitis presenting with focal seizure and relapsing lymphocytic hypophysitis. BMC Endocr Disord. 2015; 15: 64.
- Bando H, Iguchi G, Fukuoka H, Taniguchi M, Yamamoto M, Matsumoto R, et al. The prevalence of IgG4-related hypophysitis in 170 consecutive patients with hypopituitarism and/or central diabetes insipidus and review of the literature. European Journal of Endocrinol. 2013; 170: 161-172.
- Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001; 344: 732-738.
- Stone JH, Zen Y & Deshpande V. IgG4-related disease. N Engl J Med. 2012; 366: 539-551.
- Sato Y, Ohshima K, Ichimura K, Sato M, Yamadori I, Tanaka T, et al. Ocular adnexal IgG4-related disease has uniform clinicopathology. Pathol Int. 2008; 58: 465-470.
- Klepeis VE, Deshpande V, Sohani AR, et al. Sclerosing inflammatory pseudotumour of the orbit: an IgG4-related disease (Abstr). Mod Pathol. 2010; 23: 307.
- Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol and Metab. 2011; 96: 1971-1980.
- Gutenberg A, Hans V, Puchner MJA et al. Primary hypophysitis: clinicalpathological correlations. European Journal of Endocrinology. 2006; 155: 101-107.
- Cheuk W, Chan JKC. IgG4-related sclerosing disease. A critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol. 2010; 17: 303-332.