
Research Article
J Drug Discov Develop and Deliv. 2024; 10(1): 1050.
Design, Synthesis, In Silico Docking Prediction of Novel Triazolyl Tetrazole Derivatives: Evaluation of Anticancer and SIRT1 inhibition Activity
Veera Sankara Pavan Kumar Gudapati1,2; Sunder Kumar Kolli1,3*; Tejeswara Rao Allaka4*; Pilli Veera Venkata Nanda Kishore5; Mutyalanaidu Ganivada6; Mohd Afzal7; Durga Venkata Phani Kumar Gudapati1
1Department of Chemistry, BEST Innovation University, Gownivaripalli, India
2Department of Chemical Research, Cohance Life Sciences Ltd, India
3Department of Chemistry, Annamacharya Institute of Technology and Science Hyderabad, India
4Centre for Chemical Sciences and Technology, University College of Engineering, Science and Technology Hyderabad, Jawaharlal Nehru Technological University Hyderabad, India
5Chemistry Division, Department of Sciences and Humanities, VFSTR (Deemed to be University), India
6Advanced Centre of Research in High Energy Materials, University of Hyderabad, India
7Department of Chemistry, College of Science, King Saud University, Saudi Arabia
*Corresponding author: Sunder Kumar Kolli Department of Chemistry, BEST Innovation University, Gownivaripalli, Gorantla, Andhra Pradesh‒515231, India; Tejeswara Rao Allaka, Centre for Chemical Sciences and Technology, University College of Engineering, Science and Technology Hyderabad, Jawaharlal Nehru Technological University Hyderabad, Hyderabad, Telangana–500085, India. Email: sunderkolli@gmail.com; tejajntuh18@gmail.com
Received: March 18, 2024 Accepted: April 29, 2024 Published: May 06, 2024
Abstract
The development of novel techniques for modifying molecules with 1,2,3‒triazole and tetrazole scaffolds provides an intriguing basis for this research, since these compounds are important in organic synthesis, especially the production of bioactive organic compounds. In order to facilitate the creation of a more effective technique for converting anticancer heterocyclics, a new class of 1,2,3‒triazolyl tetrazoles fused with tetrahydro cyclo hepta pyridinone was created and produced. The newly created scaffolds were all characterized using mass spectrometry, FT‒IR, and NMR (1H & 13C). Since 1,2,3‒triazole and tetrazole scaffolds are important in organic synthesis, especially in the production of bioactive organic molecules, the development of new techniques for altering these compounds provides an intriguing basis for this work. A unique series of tetrahydro cyclo hepta pyridinone fused 1,2,3‒triazolyl tetrazoles was devised and synthesized to aid in the development of an effective approach for the conversion of anticancer heterocyclics. FT‒IR, NMR (1H & 13C), and mass spectrometry were used to characterize all of the recently created scaffolds. When tested against the MCF-7 cancer cell line, hybrids 8c, 8g, and 8i showed remarkable anticancer susceptibilities (IC50 = 4.5 ± 3.1, 9.2 ± 1.5, and 5.9 ± 0.7 μM). Conversely, duel heterocyclics 8a and 8c demonstrated superior anticancer inhibitory potency against the MDA-MB-231 breast cancer cell line (IC50 values 2.1 ± 1.0, 3.4 ± 1.5 μM), when compared to DXN [IC50 = 3.78 ± 0.3 μM]. Initially, all of the synthesized compounds were evaluated in silico against the target protein, hSIRT1, which suggested that compounds 8c and 8h, as well as a few additional triazole compounds, might be inhibitors. Based on docking results, 8c showed that the amino acids Val412(A), Asn346(A), Asp348(A), Phe273(A), Ile316(A), His363(A), Ala262(A), Pro318(A), Gly319(A), Tyr317(A), Gln320(A), and Glu315(A) exhibited highly stable binding to hSIRT1 receptor (PDB: 4I5I). Moreover, these scaffolds physicochemical characteristics, filtration molecular properties, assessment of toxicity, and bioactivity scores were assessed in relation to ADME (absorption, distribution, metabolism, and excretion).
Keywords: Anticancer activity; 1,2,3-triazole; Tetrazole; Pyridine; SIRT1; In silico study
Introduction
The term "Cancer" refers to a group of chronic, noncommunicable diseases linked to uncontrollably growing aberrant cells that invade and spread to neighbouring tissues, impairing physiological conditions and ultimately causing the body's vital organs to malfunction [1-3]. As of 2020, the American Cancer Society reports that cancer is the second greatest cause of mortality worldwide, accounting for 19.3 million new cases and about 10 million deaths worldwide [4]. It is projected that there will be 16.4 million deaths and about 29.5 million newly diagnosed cases by 2040 [2]. The most prevalent disease and the second most deadly kind of cancer in women is breast cancer [5]. One subtype of breast cancer known as triple-negative Breast Tumors (TNBCs) is identified by the absence of expression of the Human Epidermal growth factor Receptor-2 (HER- 2), Progesterone Receptor (PR), and Estrogen Receptor (ER) [6]. According to GLOBOCAN 2020 forecasts, cancer ranks as the second leading cause of death globally [7]. In 2020, there will be 19.3 million new instances of cancer worldwide, of which women will make up 11.7%, according to the most recent estimates of the cancer burden [8]. As such, ongoing attempts are made to satisfy the demands of the hunt for novel classes of anticancer medications.
Histone deacetylases or HDACs, are known to catalyze the removal of acetyl groups from ε-N-acetyl lysine residues in histones and nonhistone proteins [9]. This causes the DNA to coil around itself more firmly. Sirtuins are the human homologs of yeast Sir 2 (Silent information regulators 2), one of the four classes of HDACs that are conserved from yeast to humans along with I, II, III, and IV [10]. Class III HDACs called sirtuins catalyze the deacetylation process if nicotinamide adenine dinucleotide [NAD(+)] is available as a cofactor [11]. Different regions of the cell, including the cytoplasm, mitochondria, nucleus, and SIRT1, 6, and 7, contain different sirtuins [12]. Distinct N- and C-terminal sequences, as well as highly conserved NAD-binding and catalytic core domains, serve as markers for all of them [13]. Of all the sirtuins, SIRT1 has attracted the most study interest, and it has been noted that SIRT1 is involved in cancer [14]. As a result, studies have been done on SIRT1 as a potential pharmacological target for the creation of anti-cancer drugs [15]. The need for new approaches to address the complex issue of drug resistance [16], which frequently contributes significantly to chemotherapy failure, and (ii) lessen the toll that this disease takes on one's finances, social life, and personal relationships is why the SIRT1-based strategies have garnered so much attention [17]. Tumor suppressor protein p53 is a substrate of SIRT1 and also has additional substrates like Ku70, NF-jB, fork head proteins, etc., which increased the amount of p53 in response to SIRT1 inhibition, according to research [18]. As a result, research using small compounds has been done to find SIRT1 inhibitors [19].
In pharmacology, industrial materials, biology, and medicine, the tetrazole motif is a crucial synthetic scaffold that has several applications [20]. In an effort to create novel pharmacological entities, a large number of unique heterocyclic compounds have been investigated for long-term toxicity against a variety of disorders. The potential medical applications of nitrogen-containing heterocycles in tetrazole-based drugs for anticancer [21], anti-HIV [22], antiproliferative [23], antibacterial [24], antifungal [25], antitubercular [26], antihypertensive, anti-allergic, and antileishmanial activities [27]. 1,2,3-triazole and imidazole are the fundamental building blocks of many pharmaceuticals, and interest in medicinal and pharmaceutical chemistry has been piqued by these analogs. Researchers are interested in lead compounds made of 1,2,3-triazoles with heterocycles because they have a wide range of biological properties, including the ability to form dipole–dipole bonds with various enzymes, proteins, and receptors as well as antibacterial, antifungal, anticonvulsant, anti–HIV, antituberculosis, anticancer, antiviral, anti–diabetic, anti–inflammatory, and analgesic effects [28-36]. Because azides and alkynes are easy to assemble into a single structure, this reaction is beneficial in the synthesis of 1,2,3–triazoles, which are heterocyclic compounds with good yield. CuAAC (Scheme 1) is a copper (I) and azide-alkyne catalyzed cycloaddition with a variety of uses in the biological and material sciences, such as DNA synthesis, drug discovery, and oligonucleotide synthesis [37].
Pharmacochemical, pharmacokinetic, and pharmacodynamic elements have become some of the most crucial steps in the drug discovery process over the last few decades. Drug development and research organizations have developed the ability to compute characteristics in vitro in an orderly manner, with the goal of understanding the behavior of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) in vivo. In silico ADMET forecasting is expected to maximize screening and trials while lowering the likelihood of attrition from late-stage drug development procedures by concentrating on the most promising drug candidates. By mimicking the interaction of an aspirant ligand with a macromolecule (receptor), a computational modeling technique known as molecular docking predicts the optimal orientation for binding one substance to another to form a stable result. Consequently, during the medication development process, the docking technique has a major effect on the rational drug design process. Using pharmacophore modeling, molecular docking, in silico ADME, and toxicity studies, we created 1,2,3-triazole hybrids based on tetrazoles in order to understand and interpret the mechanism of binding interactions between our prepared duel heterocyclic ligands and the crystallographic SIRT1 receptor based on HDAC. In conclusion, this research has shown that possible SIRT1 inhibitors with beneficial pharmacokinetic properties exist.
Materials and Methods
Without additional purification, commercial vendors provided the starting components, which were then used. Using an Electrothermal device, all melting points were determined. Utilizing a Perkin–Elmer 100 infrared spectrophotometer with cm-1 precision, 400–4000 cm-1 of IR spectra were recorded in KBr pellets (ѵ in cm-1). Using a Bruker Avance spectrometer, the NMR spectra were captured in CDCl3 at 400 MHz for 1H and 100 MHz for 13C. Chemical changes for 1H and 13C are expressed on the δ scale (ppm) and are correlated with internal TMS. The following acronyms represent the signal multiplicities: s for singlet, d for doublet, t for triplet, q for quadruplet, and m for multiplet. The coupling constants are given in Hertz (Hz). All the reactions were monitored by Thin-Layer Chromatography (TLC) on Silica Gel 60 F254 plates; visualization by UV detection at 254 nm and were carried out under nitrogen atmosphere in dry solvents under anhydrous conditions.
Synthesis of Dimethyl 5,9-dihydroxy-7H-cyclohepta[b]pyridine-6,8-dicarboxylate (2)
Following a -5˚C cooling of the reaction, 2.39g (20.11 mmol) of thionyl chloride were added to a 2.8g (16.76 mmol) solution of pyridine-2,3-dicarboxylic acid in 28 mL methanol, and the combination refluxed for 7 hours. As demonstrated by TLC, excess methanol was distilled off once the reaction was completed.
Ethyl acetate was used to extract the residue after it had been diluted with a 10% NaHCO3 solution. After that, it was vacuum-evaporated, dried over Na2SO4, and produced a white solid that was 90% pyridine-2,3-dicarboxylic acid dimethyl diester with a melting point of 103°C. After being created, 2.5g (12.82 mmol) of dimethyl pyridine-2,3-dicarboxylate 1 were dissolved in 15 mL of toluene. Potassium tert-butoxide (1.57g, 14.10 mmol) was then gradually added at a temperature lower than 15˚C. [4 + 3] cycloaddition of mixture of 1 and Dimethyl Glutarate (DMG) (1.78 g, 15.38 mmol) in toluene (15 mL) was added to the reaction at the same temperature, and then the reaction mixture was refluxed for 10 h. Excess toluene was distilled off, the residue was poured into ice water, and the resulting solution was extracted with ethyl acetate (2 x 25 mL). The combined organic extracts were dried over Na2SO4 and evaporated under a vacuum. The crude product was purified by recrystallization from ethanol to obtain 2 as white color solid (81%), mp 114°C.
Synthesis of 7,8-Dihydro-5H-cyclohepta[b]pyridine-5,9(6H)-dione (3)
In the presence of 20mL of water at 75˚C, a mixture of dimethyl 5,9-dihydroxy-7H-cyclohepta[b]pyridine-6,8-dicarboxylate (2) (2.5g, 8.5 mmol) and HCl (0.46 g, 12.88 mmol) was heated for 10 hours. The reaction was monitored by TLC (ethyl acetate: hexane, 8:2), and once it was completed, the desired product was produced. After distilling out excess toluene, the residue was placed in ice water, and ethyl acetate (2 x 25mL) was used to extract the resultant solution. After being dried on Na2SO4, the mixed organic extracts evaporated in a vacuum. The crude product was refined by recrystallization from ethanol, yielding white solids (81%) with a melting point of 114 °C.
Synthesis of (R)-9-Hydroxy-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-5-one (4)
To the mixture of the substrate 7,8-dihydro-5H-cyclohepta[b]pyridine-5,9(6H)-dione 3 (0.02 mol, 2.5g), Rh-(R-binapine) (COD)BF4 (0.7 g, 0.009 mol) was added in 30 mL of dichloromethane at below 5˚C. The reaction was monitored at rt for 24 h, and after completion of the reaction was cooled to room temperature. The solution was diluted with 50 mL of water and then stirred overnight at room temperature in a sealed tube. After 24 h, the solution was extracted with diethyl ether (25mL x 2) following the organic layers were combined, dried over MgSO4, filtered through a cotton layer and concentrated in vacuo to give the corresponding hydrogenated product 4 (52%).
Synthesis of (R)-5-oxo-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-9-yl 4-methyl benzene sulfonate (5)
(R)-9-hydroxy-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-5-one (4) (2.2g, 12.42 mmol) in dichloromethane (15mL) under nitrogen environment, and triethylamine (1.88g, 18.63 mmol) was added at 0°C. The mixture was then stirred at room temperature for an entire night after a dropwise addition of p-tolyl sulfonyl chloride (2.60g, 13.67 mmol) in DCM (10 mL). The liquid used in the process was diluted using dichloromethane and then cleaned using 10% aq. HCl (10 mL), saturated NaHCO3 solution (10 mL), water (10 mL), brine (15 mL), and anhydrous sodium sulphate. A pale-yellow oil was obtained by separating and evaporating the organic layer. This oil was then refined using flash chromatography over silica gel and petroleum ether–acetone (1:1), yielding 5 (90%) as a white solid (mp: 85–87°C).
Synthesis of (S)-9-Azido-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-5-one (6)
(R)-5-oxo-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-9-yl 4-methylbenzene sulfonate (5) (2.0g, 6.02 mmol) was taken in anhydrous DMF (16 mL) under nitrogen atmosphere. After adding sodium azide (0.70g, 10.83 mmol) to the reaction mixture, it was stirred at 70°C for 6 h. After the reaction was finished, the reaction mass was split into 25 mL of water and 20 mL of EtOAc. The organic phase was washed with water and 0.1 M HCl (20 mL) after the layers were separated. Subsequently, the organic layer was isolated, allowed to dry on MgSO4, and allowed to evaporate at low pressure until it was totally dry. The resulting oily residue was chromatographed using hexane–EtOAc (4:1), producing 6 (86%) as a yellow oil that solidified at 144–146°C after standing.
General Procedure for the Synthesis of 8a-k
The first step in the synthesis of a series of 1-phenyl-5-(prop-2-yn-1-ylthio)-1H-tetrazoles 7a–k was used from phenyl isothiocyanate. The solution containing 1-phenyl-1H-tetrazole-5-thiol (2.1 mmol), propargyl bromide (1.2 mmol), and tetrabutylammonium bromide was then agitated for 4 hours at room temperature using a combination of triethylamine (4 mL) and DCM (6 mL). When the reaction (TLC) was complete, the reaction mixture was added to 15 mL of ice-cold water. The solid product was then filtered off, dried, and purified using column chromatography with ethyl acetate–hexane (2:8) (87%). The important intermediate 6 (2.0g, 9.85 mmol) and o-hydroxyphenyl tetrazole 7a (3.19g, 13.79 mmol) were added to DMF: water (3:2, 20 mL). Copper sulphate pentahydrate (1.96g, 7.8 mmol) and sodium ascorbate (1.56g, 7.8 mmol) were added to this reaction in addition. The reaction was held below 10 C for 20 minutes, and then it was stirred for 1 hour at room temperature. Following TLC monitoring of the reaction's development (EtOAc: Hexane 4:6), the reaction was gradually added to 200 mL of ice water and swirled. The solid products were separated with EtOAc (2 x 75 mL), filtered, thoroughly washed with water, and treated with brine solution. Upon vigorously shaking the mixture, a solid yellow precipitate developed. To obtain compound, this precipitate was filtered out and dried. In column chromatography on silica gel, a mixture of 5–10% ethyl acetate in hexane was utilized to purify the residue and yield (S)-9-(4-(((1-(2-hydroxyphenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-5-one (8a).
(S)-9-(4-(((1-(2-hydroxyphenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-5-one (8a)
1H NMR (400 MHz, CDCl3, δ, ppm): 9.68 (brs, 1H, OH), 8.16 (d, 1H, J = 8.9 Hz, Ar-H), 7.60 (d, 1H, J = 8.9 Hz, Ar-H), 7.51 (t, 1H, J = 9.3 Hz, Ar-H), 7.36 (s, 1H, triazole H), 7.29 (t, 1H, J = 9.3 Hz, Ar-H), 7.25 (d, 1H, J = 8.9 Hz, Ar-H), 7.20 (d, 1H, J = 8.9 Hz, Ar-H), 6.91 (d, 1H, J = 8.9 Hz, Ar-H), 5.02 (s, 2H, CH2), 4.70 (m, 1H, CH), 2.79 (m, 1H, CH), 2.58 (t, 1H, J = 9.9 Hz, CH), 2.19 (t, 1H, J = 9.9 Hz, CH), 1.93 (m, 1H, CH), 1.58 (m, 1H, CH), 1.48 (m, 1H, CH). 13C NMR (100 MHz, CDCl3, δ, ppm): 191.24, 167.48, 156.12, 151.95, 145.80, 138.52, 136.29, 130.58, 130.21, 129.18, 127.35, 127.22, 124.73, 124.45, 122.42, 68.31, 41.67, 37.03, 32.59, 24.55. IR (KBr, cm-1) υ: 3422.47 (‒OH), 2924.57, 2982.23 (‒CH), 1745.36 (C=O), 1593.42 (C=C), 1430.07 (C=N), 1246.67 (CSC). HRMS (m/z): 435.2440 [M + H]+. Elemental analysis for C20H18N8O2S: calcd, C, 55.29; H, 4.18; N, 25.79; S, 7.31; found, C, 55.36; H, 4.36; N, 25.93; S, 7.46. General experimental details of biological evaluation, docking techniques, spectral analysis of remaining compounds (8b-k) and spectral copies of 1H-NMR, 13C-NMR, IR and mass spectrums are included in supporting information (Figure S1-S43).
Results and Discussion
Design and Synthesis
Scheme 1, Table 2 shows the synthesis approaches used to acquire the unique target molecules. Pyridine-2,3-dicarboxylic acid and thionyl chloride reacted in methanol for 10 hours to produce the desired and good yields of dimethyl pyridine-2,3-dicarboxylate 1. [3 + 4] Compound 3 was produced in an 81% yield by cycloaddition between dimethyl pyridine-2,3-dicarboxylate 1 and Dimethyl Glutarate (DMG), which was followed by a decarboxylic reaction aided by hydrochloric acid. Furter, the known dione 3 should be readily accessible on large scale via a Dieckmann cyclization decarboxylation sequence starting from the readily available dimethyl 2,3-pyridinedicarboxylate. Following our synthetic strategy, we expected that an enantioselective ketone reduction would be complicated by the necessity to differentiate the two ketones present in compound. As such, initial efforts were focused on an enzymatic approach, and high throughput screening efforts identified Rh-(R-binapine) (COD)BF4 enzyme that reduced 7,8-dihydro-5H-cyclohepta[b]pyridine-5,9(6H)-dione (3) (0.02 mol) to the corresponding alcohol with encouraging chemo- and enantioselectivity. Gratifyingly, when the reaction using the reductase enzyme Rh-(R-binapine) (COD)BF4 was run at 2˚C in 30 mL of DCM under H2 pressure and the reaction was monitored for overnight at rt. The solution was diluted with 20 mL of water and then stirred overnight at room temperature in a sealed tube.
After 24 h, the solution was extracted with diethyl ether following the organic layers were combined, dried, filtered and concentrated in vacuo to give the corresponding product 4 [38a, b]. (R)-9-hydroxy-6,7,8,9-tetrahydro-5H-cyclohepta[b] pyridin-5-one (4) (12.42 mmol) was added to 15 mL of DCM at 0°C in a nitrogen environment, along with 18.63 mmol of triethylamine. Subsequently, a dropwise addition of p-tolyl sulfonyl chloride (13.67 mmol) in DCM (10 mL) was made, and the resulting mixture was allowed to stir overnight at room temperature. Dichloromethane was used to dilute the reaction mixture, which was then cleaned using 10% aq. HCl (10 mL), saturated NaHCO3 solution (10 mL), water (10 mL), brine (15 mL), and anhydrous sodium sulfate before being dried. After the organic layer was divided and evaporated, a light-yellow oil was obtained. This oil was then refined using flash chromatography over silica gel and petroleum ether–acetone (1:1), yielding 5 (3.66 g, 90%) of a white solid with a melting point of 85–87°C. Sodium azide was added to the tosylate in dry DMF at a temperature of 70–75°C, resulting in a pale yellow oil that solidified after a while to form the azide intermediate 6 (SN2 reaction with sodium azide), which caused the stereo enter to invert, 86% [39a, b]. According to the literature, a series of 1-phenyl-5-(prop-2-yn-1-ylthio)-1H-tetrazoles 7a–k was created by reacting phenyl isothiocyanate with sodium azide to produce 1-phenyl-1H-tetrazole-5-thiol [40]. The solution of 1-phenyl-1H-tetrazole-5-thiol (2.1 mmol), propargyl bromide (1.2 mmol), and tetrabutylammonium bromide in a mixture of triethylamine (4 mL) and DCM (6 mL) was stirred at ambient temperature for 4 h. The reaction mixture was added to 15 mL of ice-cold water when the reaction (TLC) was finished, and the solid product was filtered off, dried, and purified by column chromatography with ethyl acetate–hexane (2:8) (87%). The last step involved creating novel 1,2,3-triazole linked tetrazole scaffolds by using Click chemistry to create the appropriate alkynes 7a-k (9.0-13.79 mmol). The azide 6a (9.85 mmol) was then obtained and added to a solution of copper sulphate pentahydrate (7.8 mmol) in 20 mL of DMF: water (3:2). Next, sodium ascorbate (7.8 mmol) was added dropwise to the mixture under a nitrogen atmosphere at 10‒15°C and the reaction was then stirred for 1 h to 1.5 h at rt. The progress of reaction was checked using TLC (EtOAc: Hexane 4:6) and then gradually poured onto ice water (200 mL) with stirring. The solid products were separated by EtOAc (2 x 75 mL), fi ltered and washed thoroughly with water followed by brine solution. The mixture was stirred vigorously to form a yellow solid precipitate, which was fi ltered off and dried to afford compound. The residue was purifi ed by column chromatography on silica gel using a mixture of 5–10% ethyl acetate in hexane to obtained (S)-9-(4-(((1-(2-hydroxyphenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-6,7,8,9-tetrahydro-5H-cyclohepta[b] pyridin-5-one (8a).
We began our study on the model reaction of (S)-9-azido-6,7,8,9-tetrahydro-5H-cyclohepta[b] pyridin-5-one (6) using modified CuAAC seminal reaction conditions (Table 1), i.e., CuSO4. 5H2O, sodium ascorbate (Na(asc)) as a reducing agent for the (re)formation of reactive Cu(I) species, different solvent system to ensure adequate solubility of organic components. For comparison and to ensure triazole formation, an elevated temperature (45 to 50°C) was used based on noncatalyzed thermal reaction. High loadings of the CuI catalyst (20 mol%) and PPh3 (0.8 eq) were employed for the initial screening, which was carried out under refluxing conditions. The reaction of azide (6) with alkyne (7a) proceeded with lower yield in the presence of DCM: water (5 mL) (Table 1, entry 1). The CHCl3/H2O 3:2 (v/v) solvent system with CuI/PPh3 worked similarly (Table 1, entry 1), and increasing the excess of time from 45 min to 1 h did not prove beneficial (Table 1, entry 2). While the change of solvent to chloroform (entry 2, Table 1) or dichloromethane did not improve the yield further however, the use of DMF: water (3:2) increased the yield to 60% (entry 3, Table 1). In order to reduce reaction temperature, an attempt was made to implement the framework reaction conditions in DMF at rt, which resulted in a 53% decrease in product yield (entry 4, Table 1). Replacement of CuSO4 × 5H2O with CuI resulted in a slightly increased yield, while the absence of a reducing additive sodium ascorbate to regenerate Cu(I) from Cu(II) significantly increased the yield of the reaction (entry 5, Table 1). It is known that in an ambient atmosphere with oxygen, Cu(I) species oxidize to Cu(II), and that oxidized copper(II) species can be restored to the catalytically active +1 oxidation state by sodium ascorbate. In order to achieve our objective of developing the reaction in an ambient atmosphere, Na(asc) addition turned out to be essential. Nevertheless, after 45 minutes, introducing DCM to the reaction in water while refluxing produced the necessary product in a 64 percent yield (entry 5, Table 1). This was positive, and the reaction time was extended to one hour in order to further increase the product yield. Nevertheless, employing chloroform as the solvent resulted in a somewhat notable drop in yield (entry 6, Table 1). Despite the superior yield, product 8a was prepared with CuSO4 in t-butanol to obtained 68% yield (entry 7, Table 1). Accordingly, the reaction was carried out in the presence of catalyst and without any catalyst, respectively. While the yield was decreased in the first case (entry 8, Table 1) interestingly the reaction proceeded in the presence of heating conditions too affording 8a albeit in lower yield.
![]()
S.No.
Catalyst(s)
Solvent [ratio v/v]
Temperature
time
8a(%)b
1
CuI/PPh3
DCM/H2O [3:2]
45˚C
45 min
46
2
CuI/PPh3
CHCl3/H2O [3:2]
45˚C
1 h
39
3
CuI/PPh3
DMF/H2O [3:2]
45˚C
45 min
60
4
CuI/PPh3
DMF/H2O [3:2]
rt
1.5 h
53
5
CuSO4/Na(asc)
DCM/H2O [3:2]
45˚C
45 min
64
6
CuSO4/Na(asc)
CHCl3/H2O [3:2]
45˚C
1 h
59
7
CuSO4/Na(asc)
t–BuOH/H2O [3:2]
45˚C
45 min
68
8
CuSO4/Na(asc)
DMF/H2O [3:2]
45˚C
1 h
75c
9
CuSO4/Na(asc)
DMF/H2O [3:2]
rt
1.5 h
83d
aReaction conditions: Azide (6, 2.4 mmol), alkyne (7a, 3.4 mmol), catalyst (CuI/CuSO4, 1.9 mmol), sodium ascorbate (1.9 mmol), and DMF/H2O/3:2 (v/v/, 5 mL). bYield of purified product 8a after column chromatography. cThe reaction was performed in the absence of rt. dConversion into product 8a was determined by 1H-NMR spectroscopy using TMS as an internal standard.
Table 1: Initial optimization of reaction conditionsa.
Similar observation was noted when the reaction temp was decreased to rt in the presence of DMF to give 73% yield (entry 9, Table 1). Thus, the condition of entry 9 of Table 1 was identified as optimal and was used for the synthesis of analogues of 8a. After the finalization of the reaction, DMF: water (3:2) was added to the reaction composition, and the organic layer was dried, filtered off, and washed with acetone to obtained the pure (S)-9-(4-(((1-(2-hydroxyphenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-5-one (8a). Accordingly, the methodology was explored for the preparation of a variety of pyrrolidine substituted 1,2,3–triazole derivatives (Table 2) via employing a range of substrates.
![]()
8a/83%
8b/70%
8c/66%
8d/63%
8e/82%
8f/84%
8g/79%
8h/73%
8i/85%
8j/65%
8k/70%











Table 2: List of substituted tetrahydro cyclo hepta pyridinone triazole derivatives (8) synthesized (Scheme 1).
Structural elucidation of all synthetic compounds was done using distinct spectroscopic techniques such as 1H‒NMR, 13C‒NMR, IR and mass analysis. IR spectra of 8a represented characteristic absorption bands at the regions 3422.47, 2924.57, and 2982.23 cm−1 due to ‒OH, and ‒CH groups respectively. Furthermore, 8a represented an additional band at 1745.36, 1593.42 cm−1 due to its C=O, and C=N moieties, whereas its stretching vibrations at 1430.07, and 1246.67 cm−1 confirmed the presence of −C=N, −CSC groups, respectively. 1H NMR spectra of the new compound 8a revealed singlet protons of hydroxy and 1,2,3-triazole was confirmed at δ = 9.68, 7.36 ppm, while the pyridinyl doublet protons were confirmed at δ 8.16, 7.60 ppm respectively. In addition, the aromatic multiplet signals (doublets, triplets) appeared in the corresponding range of δ 7.51-6.91 ppm. Moreover, the four multiplet signals appeared at δ 4.70, 2.79, 1.93, 1.58 ppm confirming the methylene protons. In this compound, protons were shown as a triplet peak at δ 2.58, 2.19 ppm corresponding to remaining methylene protons, a singlet peak at δ 5.02 ppm for s-methyl protons. 13C-NMR spectra of compound 8a, the represented signals at δ 191.24, 167.48, and 156.12 ppm ascribed to cyclohepta[b]pyridinone, pyridine and triazole carbons and remaining signals at the deshelding region δ 145.80-122.42 ppm referring to the aromatic carbons. Also, the methylene residues of 8a appeared as five additional singlets at δ 68.31, 41.67, 32.59, 37.03, and 24.55 ppm.
Molecular weight at m/z = 435.2440 of mass spectra is evidence of compound (S)-9-(4-(((1-(2-hydroxyphenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-5-one (8a), which were in accordance with the proposed structures representing their correct molecular ion peaks beside some other important peaks (see. Supporting information).
In vitro Cytotoxicity
The ATCC methodology was followed in order to test the finished compounds anticancer activities using the MTT colorimetric assay [41]. The produced benzimidazole-triazole derivatives (8a-k) were tested in vitro against human breast malignancies (MCF-7, MDA-MB-231) shown in Table 3 to determine their cytotoxic potential. Doxorubicin was used as a reference. Only the inhibitory concentration values of those medications demonstrated 50% inhibition (IC50), and the results are shown as the percentage of inhibition at 50 μM. Table 3 displays that out of all the triazoles that were examined, 8a-k exhibit notable anticancer activity, with IC50 values ranging from 4.5 ± 3.1–43.2 ± 3.1 μM. Compound 8a's obtained IC50 values against MCF-7 and MDA-MB-231 were 8.9 ± 1.4 and 2.1 ± 1.0 μM, respectively. The findings of the screening indicate that the triazole derivatives 5e and 5k, when produced, exhibited good cytotoxic effects on the MCF-7 breast cancer cell line, with IC50 values of 13.3 ± 2.4 and 14.6 ± 1.0 (IC50 values below 15 μM).Compounds with p-methoxyphenyl substituted tetrazole (8c), difluorophenyl substituted tetrazole (8g), o-nitrophenyl substituted tetrazole (8i) elicited excellent anticancer activity against MCF-7 cell line with IC50 values 4.5 ± 3.1, 9.2 ± 1.5, 5.9 ± 0.7 μM when compared to DXN (IC50 = 4.83 ± 0.2 μM) respectively. Morover the compounds 8d, 8f and 8h showed poor anticancer activity against MCF-7 cell line, whereas remaining triazole derivatives 8b and 8j did not showed any anticancer activity even upto concentration 50 μg/mL. In contrast 1,2,3-triazole derivatives 8a and 8c reveled highest anticancer activity against MDA-MB-231 cell line accompanied by IC50 = 2.1 ± 1.0, 3.4 ± 1.5 μM, when compared to DXN [IC50 = 3.78 ± 0.3 μM]. Novel 1,2,3‒triazolyltetrazoles 8d, 8g, and 8i revealed potent antiprofilative potency against MDA-MB-231 cell line with corresponding IC50 7.1 ± ± 0.9, 7.7 ± 1.2 and 7.2 ± 2.9 μM respectively. Compounds 8b, 8f and 8h displayed moderate anticancer activity against human breast cancer cell line (MDA-MB-231) with their IC50 values 15.2 ± 1.2, 22.2 ± 3.1, and 18.7 ± 2.3 μM, where as the target compounds 8e and 8k did not exhibited the anticancer activity against MDA-MB-231 cell line. Compounds 8a, 8c, 8g, and 8i revealed almost comparable and very prestigious anticancer activity against both MCF-7 and MDA-MB-231 cell lines.
![]()
Entry
Breast cancer cell line (IC50 in µM
Enzymatic assay
MCF-7
MDA-MB-231
% of inhibition (10 µM)
IC50 (µM)
8a
8.9 ± 1.4
2.1 ± 1.0
90.6
33.19 ± 2.01
8b
NA
15.2 ± 1.2
74.8
--
8c
4.5 ± 3.1
3.4 ± 1.5
85.2
23.10 ± 1.42
8d
19.4 ± 2.2
7.1 ± ± 0.9
69.4
--
8e
13.3 ± 2.4
NA
67.2
65.3 ± 0.91
8f
43.2 ± 3.1
22.2 ± 3.1
70.2
--
8g
9.2 ± 1.5
7.7 ± 1.2
92.5
55.50 ± 2.10
8h
25.4 ± 3.9
18.7 ± 2.3
80.4
44.21 ± 1.77
8i
5.9 ± 0.7
7.2 ± 2.9
78.1
88.30 ± 2.62
8j
NA
41.4 ± 2.7
65.2
--
8k
14.6 ± 1.0
NA
68.2
--
cDXN
4.8 ± 0.2
3.7 ± 0.3
0
--
ND
ND
--
105.2 ± 1.31
cDXN: Doxorubicin; IC50: compound concentration required to inhibit the cell viability by 50%; SEM: standard error mean; each value is the mean of three independent determinations; 1–5 (very strong). 5–10 (strong); 10–25 (moderate); 25–50 (weak), and above 50 (non-cytotoxic); NA: IC50 value above 50 µM. bData represent the mean values of three independent determinations.
Table 3: Anticancer activity of the synthesized 1,2,3-triazole derivatives against breast cancer cell linesa; SIRT1 inhibition of substituted triazoles (8) in vitrob.
Next, in addition to compounds 8c, 8g, and 8i, we concentrated on compounds 8a-b, 8d-f, 8h, and 8j-k that had estimated total energy equal to or more than 20 kcal/mol. Using a well-established biochemical enzymatic assay technique, the SIRT1 inhibitory ability of these triazole-tetrazole derivatives was evaluated in vitro [42]. The results of this assay indicated that these compounds were efficient against SIRT1, as each of them inhibited the enzyme more than 50% at a dosage of 10 μM (Table 4). Motivated by the results of in silico experiments, we evaluated the inhibitory effects of all the produced compounds against SIRT1 in vitro. All the compounds were tested at a concentration of 10 μM and the readily available inhibitor Vorinostat (with the reported IC50 value against SIRT1 = 105.3 ± 1.31 μM) was used as a positive control [43]. A majority of the compounds showed inhibition higher than 50% (Table 3) signifying the usefulness of the current series of compounds against SIRT1. In this in vitro assay, the compounds 8a, 8c, 8g, and 8i that were shown to be the most advantageous in the in silico docking studies were also found to be the best active derivatives. It was discovered that the derivatives 8e and 8h were likewise active among the remaining chemicals. Interestingly, in terms of activity, none of the pyridinyl tetrazole derivatives (8j–k) seemed to be less promising. With IC50 values of 28.9± 1.4, 35.4 ± 3.9, and 35.9 ± 0.7 μM, respectively, compounds 7a, 7h, and 7i containing isopropyl substituted triazole, difluoro phenyl substituted triazole, and mono fluoro phenyl substituted triazole evoked good to moderate activity against HCA-7 cell line.Morover the compounds 5b, 5d and 5g displayed potent anticancer activity against HT29 cell line with IC50 values 13.2 ± 1.2, 7.1 ± 0.9, 7.7 ± 1.2 μM when compared to DXN (IC50 = 6.78 ± 0.3 μM) respectively. The current series of compounds SAR (Structure-Activity-Relationship) revealed that the substituted groups at the C-11 position of the tetrahydro-5H-cyclohepta[b]pyridin-5-one ring play a crucial function. It was discovered that the most active positions at this position were the p-methoxyphenyl ring, the o, p-difluoro phenyl ring, and finally the 2-nitro phenyl moiety. The inclusion of various aryl moieties investigated at this location typically yielded compounds with moderate or low activity, while p-methyl or p-chloro phenyl substituted tetrazoles at the same position seemed to be tolerated. Actually, a small alteration to the intended compounds caused a significant shift in their activity, indicating that compounds' activity rose as atoms' tendency to remove electrons increased. It was discovered that the nitrogen atom was less advantageous than the carbon atom (8j and 8k). However, the IC50 values found for the most active compounds, as shown in Table 3 and Figure 1, supported the results of the original in vitro tests that were followed by in silico research. Among the compounds studied, compound 8g was shown to be the most potent, while compounds 8c and 8i proved to be the next best. Additionally, it appeared that all three of these compounds, as well as 8a, 8e, and 8h, were many times more effective than vorinostat.
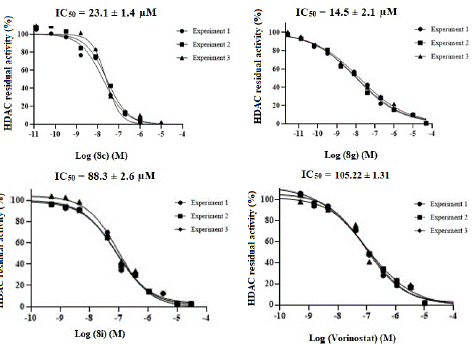
Figure 1: IC50 of compounds 8c, 8g, 8i and Vorinostat on HDAC inhibitors. Data are expressed as means of the three experiments ± SD.
![]()
Ligand
Hydrophobic interactions
Residue (H-bond type)Length (Å)
Hydrophobic interactions
Residue (bond type)Bond energy/ Ki
8c
Val412(A) [H-bond]
Asn346(A) [H-bond]
Asp348(A) [C-H bond]
Phe273(A) [C-H bond]2.98 Å
2.78 Å
2.84 Å
3.05 ÅIle316(A), Ile270(A), Ile347(A), Asn346(A) [π-sigma]
His363(A), Ile411(A), Ile279(A), Ala262(A), Phe273(A), Ile347(A), Ile316(A) x 2 [π-alkyl]
Phe413(A), Pro318(A), Gly319(A), Tyr317(A), Gln320(A), Glu315(A), Phe297(A), Gln345(A), Pro271(A) [Van der Waals]-11.79/2.29 nM
8g
Val412(A) [H-bond]
Asn346(A) [H-bond]
Ile347(A) [H-bond]
Phe273(A) [C-H bond]2.95 Å
2.83 Å
2.80 Å
3.16 ÅIle270(A), Ile279(A), Asn346(A) [Π-sigma]
Ile347(A), Ile316(A), Glu315(A) [Halogenated]
Phe273(A) x 2 [π-π]
Ala262(A), His363(A), Ile411(A), Ile347(A) X 2, Ile316(A) x 2, Phe273(A) [π-alkyl]
Phe413(A), Gln320(A), Phe297(A), Phe321(A), Asp348(A), Pro271(A), Gln345(A) [Van der Waals]-11.26/5.59 nM
8i
Val412(A) [H-bond]
Asn346(A) [H-bond]
Phe273(A) [C-H bond]
Pro271(A) [C-H bond]
Ile316(A) [C-H bond]
Ile270(A) [C-H bond]
Ile347(A) [C-H bond]2.82 Å
2.66 Å
2.85 Å
2.56 Å
3.06 Å
2.75 Å
3.01 ÅAsn346(A), Ile270(A), Ile347(A), Ile316(A) [π-sigma]
Phe273(A) [π-π]
Ala262(A), His363(A), Ile279(A), Ile316(A) x 2, Ile270(A), Ile411(A), Ile347(A) [π-alkyl]
Gln345(A), Glu315(A), Phe321(A), Gln320(A), Asp348(A), Pro318(A), Phe297(A), Phe413(A), Tyr317(A) [Van der Waals]-12.66/522.32 µM
Table 4: Molecular interactions of ligands with amino acids of HDAC binding protein of hSIRT1 receptor (PDB: 4I5I).
Autodock Binding Affinities of the Synthesized Compounds into hSIRT1 Receptor HDAC
Once the library of triazole linked to tetrahydro-5H-cyclohepta[b]pyridine derivatives (8) was successfully accessed quickly under CuAAC conditions, these compounds were first evaluated in silico against the target protein, hSIRT1. Using computational and experimental methodologies together is a compelling approach to designing and optimizing effective medication candidates. In order to do docking investigations of these drugs, the crystal structure of hSIRT1 coupled to an HDAC inhibitor (PDB: 4I5I) was used. For this, a graphical automatic drug design system, or the autodock4.2 version software, which is well-known to be appropriate for docking, screening, and analysis, was used [44-46]. Based on the estimated total energy of compounds presented in Table 4, the 1,2,3-triazoles 8c, 8g, and 8i were considered as the most promising and potential hits that seemed to be better than known SIRT1 inhibitor Vorinostat. Besides, a number of other compounds showed estimated total energy equal to or greater than 10 kcal/mol that include 8a-b, 8d-f, 8h, and 8j-k. This was demonstrated by the docked pose's ability to replicate all of the crucial interactions that the co-crystallized ligands achieved with the hot spots in the active sites, including Val412(A), Asn346(A), Ile347(A), Phe273(A), and His363(A). Additionally, the small RMSD between the docked pose and the co-crystallized ligand was 0.69 Å. Up to three hydrogen bonds were detected between the docked compounds and hSIRT1 receptor HDAC. The formed hydrogen bonds are established within distances of 2.56–3.16 Å and binding free energies (ΔG) revealed were in the range of (‒11.26 to ‒12.66 kcal/mol), with corresponding inhibition constants (Ki) being 2.29 nM to 522.32 μM as represented in Table 4.
The docking of ligand 8c within the histone deacetylase pocket of hSIRT1 receptor allowing energy score −11.79 kcal/mol, it was found that O32 of the 5-oxo-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridine and N-linker played a vital role in the binding through a monodentate hydrogen bonded interaction with the backbone of the 4I5I protein with amino acids Val412(A) (distance: 2.98 Å ). Moreover, the N15 of triazole ring shared fixation through H-bond interaction with Asn346(A) with their bond distance 2.78 Å . This p-methoxyphenyl, cyclo hepta pyridinone, and triazoles were stabilized by π‒sigma stackings with Ile316(A), Ile270(A), Ile347(A) and Asn346(A) amino acids. and through hydrophobic interactions with His363(A), Ile411(A), Ile279(A), Ala262(A), Phe273(A), Ile347(A), Ile316(A) x 2 via π‒alkyl stackings as shown in Figure 2 & 5. Compound 8c was further stabilized by van der Waals interactions of its hepta pyridinone and triazole with amino acid residues Phe413(A), Pro318(A), Gly319(A), Tyr317(A), Gln320(A), Glu315(A), Phe297(A), Gln345(A), and Pro271(A) respectively. Finally, it can be formed two carbon hydrogen bonds with the key active site of amino acids Asp348(A), Phe273(A) in the catalytic site enhance the binding affinity.
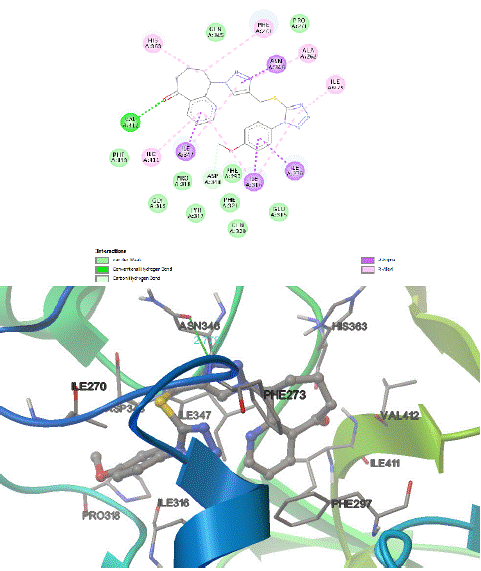
Figure 2: 2D‒ and 3D‒ schematic binding interactions of compound 8c into active site of anticancer protein 4I5I.
In addition, we also found that the binding mode predicted for 8g allow a better fit the entrance of HDAC binding site, maximizing the van der Waals interactions with Phe413(A), Gln320(A), Phe297(A), Phe321(A), Asp348(A), Pro271(A), Gln345(A) residues, and potentially justifying its increased activity compared to reference ligand. The difluoro phenyl, tetrazole and triazole rings of 8g shows hydrophobic interactions [π‒sigma] between the amino acids Ile270(A), Ile279(A), Asn346(A) respectively. While the free fluorophenyl group contains tetrazole shows three hydrophilic interactions (halogenated) with Ile347(A), Ile316(A), Glu315(A). As illustrated in the docking of 8g, the tetrazole and triazole scaffolds formed π‒π stackings with the backbones of Phe273(A) [3.04 Å ], Phe273(A) [2.99 Å ] within the hSIRT1 binding pocket. Moreover, the cyclo hepta pyridinone, triazole moieties improved fixation within 4I5I through the formation of three H-bondings with Val412(A) [O32…N], Asn346(A) [N15…OD1], Ile347(A) [N15…N] and their bond distance 2.95 A˚, 2.83 A˚ and 2.80 Å respectively. In addition, this ligand displayed a highest π‒alkyl stackings with residues Ala262(A), His363(A), Ile411(A), Ile347(A) X 2, Ile316(A) X 2, Phe273(A) in the active site of hSIRT1 receptor (Figure 3 and 5).
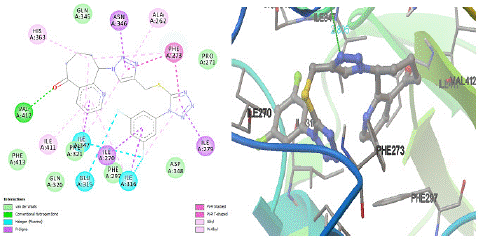
Figure 3: 2D and 3D representations of 8g conformation at the binding pocket of HDAC in hSIRT1 receptor (4I5I).
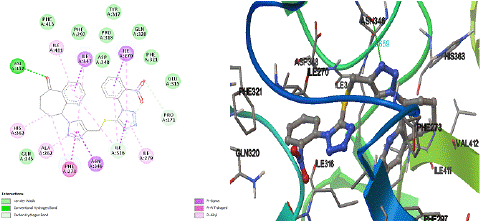
Figure 4: Two and three-dimensional interactions of 8i with the hSIRT1 binding site of HDAC (4I5I).
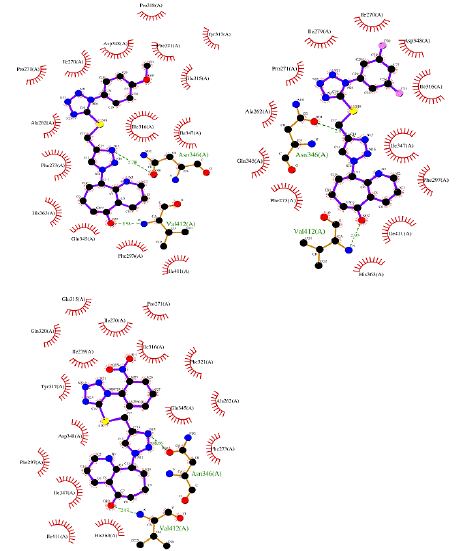
Figure 5: Ligplot interacting confirmations of compound 8c, 8g, and 8i into HDAC active site of hSIRT1 receptor (4I5I).
By focusing upon compound 8i as the most active inhibitor, it fulfilled the key interactions in the active site of hSIRT1 receptor with energy score −12.66 kcal/mol, where hydrogen bondings was established between O32 of the cyclo hepta pyridinone moiety and nitrogen of the side chain of Val412(A), and N15 of triazole and OD2 of the side chain of Asn346(A) and their bond distance 2.82 Å , 2.66 Å accordingly (Figure 4). On the other hand, the 2-nitrophenyl triazolyl tetrazole derivative 8i was found to highest van der Waals located in Gln345(A), Glu315(A), Phe321(A), Gln320(A), Asp348(A), Pro318(A), Phe297(A), Phe413(A), Tyr317(A) amino acids respectively. In addition, compound 8i formed eight Π‒alkyl bondings via residues Ala262(A), His363(A), Ile279(A), Ile316(A) x 2, Ile270(A), Ile411(A), Ile347(A) respectively. In particular, the 1,2,3-triazole ring of 8i predominantly interact with the side chain of Phe273(A) through π‒π interaction with distance 3.11 Å . Differently, the triazole, tetrazole and cyclo hepta pyridinone fragments forms five extensive carbon-H bondings with amino acid residues Phe273(A), Pro271(A), Ile316(A), Ile270(A), Ile347(A) buried within the binding cavity via 2.85 Å , 2.56 Å , 3.06 Å , 2.75 Å , and 3.01 Å distance respectively. Molecule 8i showed an additional hydrophobic interaction like π‒sigma between 1,2,3-triazole and Asn346(A) residue, tetrazole and Ile316(A), nitrobenzene and Ile270(A) pyridine and Ile347(A), which corresponds to the higher binding value and better anticancer activity when compared to DXN.
In silico ADMET Prediction of Synthesized Compounds
SwissADME online web tool provided by the Swiss Institute of Bioinformatics (SIB) was implemented for the calculation of the physicochemical properties in addition to the prediction of the pharmacokinetic properties and drug-likeness of the novel heterocyclic candidates 8a-k (Table 5 and Figure 6) [47,48]. It is anticipated that the substances submitted would have encouraging pharmacokinetic and physicochemical characteristics. Every synthetic chemical (MW, mlogp, HBA, HBD, RB, and PSA) followed the Lipinski guidelines. Nonetheless, every chemical derivative that was generated was compatible with Lipinski. Every substance examined had a MW of less than 500. The molecules are therefore tiny, portable, quickly distributed, and effectively absorbed. Due to their abundance of rotatable bonds (RBs 5–6), all compounds in this class have a significant degree of structural flexibility. Less than 10 RBs increases the likelihood that a compound will be bioavailable. The availability of more RBs will influence their ability to successfully engage with particular binding sites. To avoid having to remove compounds with bad qualities later on in the process, ideally before synthesis, it is imperative to calculate ADMET parameters early on in the creation of a medication or pesticide. The synthesized compounds (8a–k) were evaluated for lipophilicity, solubility, and Lipinski's Rule of Five predictions in order to create excellent oral medications. Some theoretical ADME predictions of the newly synthesized compounds can aid in further in vitro and in vivo drug research. Thus, ADME online tools were used to compute the specified compound features, such as pharmacokinetics, lipophilicity, drug-likeness, water-solubility, toxicity risks, and physicochemical characteristics (Table 5) [49].
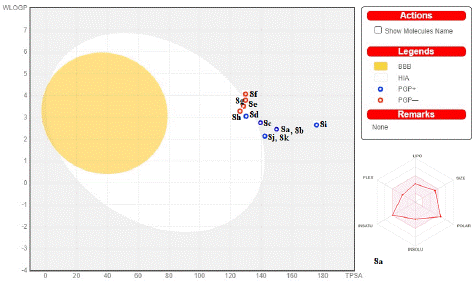
Figure 6: The boiled egg model for 8a-k. In terms of the medications being designed, white color stands for gastrointestinal absorption and yellow color for blood brain barrier (BBB). The proposed drugs are depicted by red circles.
![]()
S.No.
8a
8b
8c
8d
8e
8f
8g
8h
8i
8j
8k
MW
434.47
434.47
448.50
432.50
452.92
487.37
454.46
436.47
463.47
419.46
419.46
Heavy Atoms
31
31
32
31
31
32
32
31
33
30
30
Csp3
0.25
0.25
0.29
0.29
0.25
0.25
0.25
0.25
0.25
0.26
0.26
nRoBt
5
5
6
5
5
5
5
5
6
5
5
H-BA
8
8
8
7
7
7
9
8
9
8
8
H-BD
1
1
0
0
0
0
0
0
0
0
0
M-refractivity
112.20
112.20
116.67
115.15
115.19
120.20
110.10
110.14
119.00
107.98
107.98
TPSA (A˚2)
149.80
149.80
138.80
129.57
129.57
129.57
129.57
129.57
175.39
142.46
142.46
iLog Po/w
2.92
2.26
2.89
2.69
2.98
2.99
2.93
3.02
2.48
2.67
2.58
XLog Po/w
1.80
1.80
2.13
2.52
2.79
3.41
2.36
2.26
1.99
1.09
1.09
WLog Po/w
2.45
2.45
2.75
3.05
3.40
4.05
3.86
3.30
2.65
2.14
2.14
mLog Po/w
1.78
1.78
2.00
2.50
2.77
3.26
3.04
2.66
1.48
1.28
1.28
ESOl logs
-3.86
-3.86
-4.08
-4.30
-4.60
-5.19
-4.32
-4.17
-4.06
-3.34
-3.34
Silicos logs
-5.36
-5.36
-6.05
-6.33
-6.53
-7.11
-6.47
-6.21
-5.29
-5.58
-5.58
GI
Low
Low
Low
High
High
Low
Low
High
Low
Low
Low
BBB
No
No
No
No
No
No
No
No
No
No
No
P-gp. sub.
Yes
Yes
Yes
Yes
No
No
No
No
Yes
Yes
Yes
CYP1A2
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
CYP2C19
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
CYP2C9
No
No
Yes
Yes
Yes
Yes
No
No
No
No
No
CYP2D6
No
No
No
No
No
No
No
No
No
No
No
CYP3A4
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Skin perm.
-7.67
-7.67
-7.52
-7.15
-7.08
-6.85
-7.40
-7.36
-7.71
-8.08
-8.08
Lipinski
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Ghose
Yes
Yes
Yes
Yes
Yes
No
No
No
Yes
Yes
Yes
Veber
No
No
Yes
Yes
Yes
Yes
Yes
Yes
No
No
No
Egan
No
No
No
Yes
Yes
Yes
Yes
Yes
No
No
No
Muegge
Yes
Yes
Yes
Yes
Yes
Yes
Yes
Yes
No
Yes
Yes
Bio. Score
0.55
0.55
0.55
0.55
0.55
0.55
0.55
0.55
0.55
0.55
0.55
Lead likeness
1
1
1
1
1
1
1
1
1
1
1
Syn. Acces.
3.91
3.91
3.99
3.99
3.88
3.92
3.88
3.88
4.11
3.93
3.90
Table 5: Prediction of pharmacokinetics and physicochemical properties.
All compounds, with the exception of (8a, 8b, and 8i), would not pass the blood–brain barrier and their gastrointestinal absorption would be higher, according to Figure 6. Because it suppresses the CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4enzymes, it may also interact with other medications [50]. They showed good GIT absorption, moderate water solubility, and a projected wlogP in the range of 2.14–4.05. The drug-likeness criteria established by prominent pharmaceutical corporations, including Amgen, Ghose, Pfizer, Bailey, Veber, and Pharmacia, are met by the promising compounds. Additionally, all derivatives Log S values fall between -5.36 and -7.11, indicating a moderate water solubility. The synthesis of bioactive compounds, especially their formulation and handling, is significantly facilitated by the presence of a soluble molecule. As a result of these findings, all compounds have a high theoretical bioavailability and might be used as drug-like and anticancer agents.
Conclusion
In conclusion, we focused on the tetrahydro-5H-cyclohepta[b]pyridin-5-one framework for the design and synthesis of compounds that could potentially inhibit SIRT1. In light of this, the target compounds were conventionally synthesized utilizing the environmentally friendly alkyne-azide functionalized CuAAC coupling procedure. Their structural identity was then verified by FTIR, 1H and 13C NMR, and HRMS. To assess the effects of substituents at various positions on the biological activities against a human breast cancer cell line (MCF-7 & MDA-MB-231), pharmacological tests were conducted. Several of these 1,2,3-triazolyl tetrazole derivatives demonstrated encouraging inhibitory effects (> 50% inhibition) when tested in vitro against SIRT1, which was consistent with the findings of docking studies. Upon preliminary evaluation of the synthesized compounds against hSIRT1 in silico, multiple compounds, including 8a, 8c, 8e, 8g, and 8i, were identified as potential inhibitors. The majority of active drugs' docking posture contacts with the hSIRT1 receptor's HDAC active site (4I5I) revealed three significant H-bond interactions with the protein's residues, Val412(A), Asn346(A), Ile347(A) which were mirrored in their estimated total energy. However, our efforts, which included the original design, conventional synthesis, in vitro and in silico tests, made it easier to identify tetrahydro-5H-cyclohepta[b]pyridin-5-one based triazolyl tetrazoles as putative inhibitors of SIRT1 for additional research. Thus, we have revealed the potential therapeutic utility of this class of compounds against cancer as well as the efficiency of copper catalyst for the synthesis of tetrahydro-cyclohepta pyridinone derivatives.
Highlights
• 1,2,3‒Triazolyl tetrazoles fused with tetrahydro cyclo hepta pyridinones were created and synthesised on the basis of virtual screening.
• In vitro studies of the synthesized compounds were evaluated utilizing MTT colorimetric assay using MCF-7, MDA-MB 231 cell lines.
• Safety profiles of the most potent derivatives were screening into hSIRT1 receptor on HDAC inhibitors.
• Computational molecular modeling, docking energy calculations and ADMET predictions were performed to know the best lead candidates.
• In comparison to the reference drug doxorubicin, the compound 8c displayed good in-vitro activities for all the parameters.
Author Statements
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Author Contributions Statement
Mr. Gudapati Veera Sankara Pavan Kumar, Dr. Kolli Sunder Kumar, Dr. A. Tejeswara Rao, Prof. Mohd Afzal in charge of write up the paper, synthesis, biological evaluation, and the computational docking calculations. Dr. PVVN Kishore, Dr. G. Mutyalanaidu, Mr. Durga Venkata Phani Kumar Gudapati for review the paper and analytical techniques.
Acknowledgements
The authors (Gudapati) are thankful to Cohance life sciences Ltd & Bharatiya Engineering Science & Technology Innovation University for providing required facilities for completion of the research work. We also extend our gratitude towards Osmania University, University of Hyderabad, Hyderabad for providing molecular docking and biological studies. The authors extend their appreciation to Researchers Supporting Project number (RSPD2024R979), King Saud University, Riyadh, Saudi Arabia.
Data Availability
The data used in this study are available from the corresponding author upon interest.
Supporting Information
General experimental details of biological activity evaluation, computational docking studies and copies of 1H-NMR, 13C-NMR, Mass and IR spectrums are included in supporting information (Figure S1-S43). The data that support the findings of this study are available in the supplementary material of this article.
References
- Hsieh CY, Ko PW, Chang YJ, Kapoor M, Liang YC, Chu HL, et al. Design and synthesis of benzimidazole-chalcone derivatives as potential anticancer agents. Molecules. 2019; 24: 3259.
- Satija G, Sharma B, Madan A, Iqubal A, Shaquiquzzaman M, Akhter M, et al. Benzimidazole based derivatives as anticancer agents: Structure activity relationship analysis for various targets. J Heterocycl Chem. 2022; 59: 22−66.
- Kelishadi R, Farajian S. The protective effects of breastfeeding on chronic non-communicable diseases in adulthood: A review of evidence. Adv Biomed Res. 2014; 3: 3.
- Cao W, Chen HD, Yu YW, Li N, Chen WQ. Changing profiles of cancer burden worldwide and in China: a secondary analysis of the global cancer statistics 2020. Chin Med J. 2021; 134: 783−791.
- Giaquinto AN, Sung H, Miller KD, Kramer JL, Newman LA, Minihan A, et al. Breast cancer statistics, 2022. CA Cancer J Clin. 2022; 72: 524−541.
- Stevens KN, Vachon CM, Couch FJ. Genetic susceptibility to triple-negative breast cancer. Cancer Res. 2013; 73: 2025−2030.
- Sung H, Ferlay J, Siegel RL, Global Cancer Statistics 2020 GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021; 71: 209–249.
- Chhikara BS, Parang K. Global Cancer Statistics 2022: the trends projection analysis. Chem Biol Lett. 2023; 10: 451.
- Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014; 6: a018713.
- North BJ, Verdin E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004; 5: 224.
- Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007; 404: 1-13.
- Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000; 273: 793-798.
- Yuan H, Marmorstein R. Structural basis for sirtuin activity and inhibition. J Biol Chem. 2012; 287: 42428–42435.
- Liu T, Liu PY, Marshall GM. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009; 69: 1702-1705.
- Jeong SM, Haigis MC. Sirtuins in cancer: a balancing act between genome stability and metabolism. Mol Cells. 2015; 38: 750-758.
- Wang Z, Chen W. Emerging Roles of SIRT1 in Cancer Drug Resistance. Genes Cancer. 2013; 4: 82-90.
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65: 87–108.
- DellOmo G, Crescenti D, Vantaggiato C, Parravicini C, Borroni AP, Rizzi N, et al. Inhibition of SIRT1 deacetylase and p53 activation uncouples the anti-inflammatory and chemopreventive actions of NSAIDs. Br J Cancer. 2019; 120: 537-546.
- Spinck M, Bischoff M, Lampe P, Meyer-Almes FJ, Sievers S, Neumann H. Discovery of Dihydro-1,4-Benzoxazine Carboxamides as Potent and Highly Selective Inhibitors of Sirtuin-1. J Med Chem. 2021; 64: 5838-5849.
- Wei CX, Bian M, Gong GH. Tetrazolium Compounds: Synthesis and Applications in Medicine. Molecules. 2015; 20: 5528−5553.
- Constantnos GN, Ting Z, Alexander D. Tetrazoles via Multicomponent Reactions. Chem Rev. 2019; 119: 1970–2042.
- Zhan P, Li Z, Liu X, De Clercq E. Sulfanyltriazole/tetrazoles: a promising class of HIV-1 NNRTIs. Mini Rev Med Chem. 2009; 9: 1014–1023.
- Dileep K, Polepalli S, Jain N, Buddana SK, Prakasham RS, Murty MSR. Synthesis of novel tetrazole containing hybrid ciprofloxacin and pipemidic acid analogues and preliminary biological evaluation of their antibacterial and antiproliferative activity. Mol Diversity. 2018; 22: 83–93.
- Siva Kumar Gandham, Amit A Kudale, Tejeswara Rao Allaka, Kalyani Chepuri, Anjali Jha. New indazole–1,2,3–triazoles as potent antimicrobial agents: Design, synthesis, molecular modeling and in silico ADME profiles. Journal of Molecular Structure. 2024; 1295: 136714.
- Gujja V, Sadineni K, Epuru MR, Rao Allaka T, Banothu V, Gunda SK, et al. Synthesis and in Silico Studies of Some New 1,2,3-Triazolyltetrazole Bearing Indazole Derivatives as Potent Antimicrobial Agents. Chem Biodiversity. 2023; 20: e202301232.
- Chauhan K, Sharma M, Trivedi P, Chaturvedi V, Chauhan PM. New class of methyl tetrazole based hybrid of (Z)-5-benzylidene-2-(piperazin-1-yl) thiazol-4 (%H)-one as potent antitubercular agents. Bioorg Med Chem Lett. 2014; 24: 4166–4170.
- Purohit P, Pandey AK, Singh D, Chouhan PS, Ramalingam K, Shukla M, et al. An insight into tetra hydro-β-carboline–tetrazole hybrids: synthesis and bio evaluation as potent antileishmanial agents. Med Chem Commun. 2017; 8: 1824–1834.
- Ananda Kumar D, Tejeswara Rao A, Yugandhar K, Sunil Kumar N, Pradeep P, et al. Design, synthesis and biological evaluation of novel substituted indazole-1,2,3-triazolyl-1,3,4-oxadiazoles: Antimicrobial activity evaluation and docking study. Results in Chemistry. 2022; 4: 100605.
- Bonandi E, Christodoulou MS, Fumagalli G, Perdicchia D, Rastelli G, Passarella D. The 1,2,3-triazole Ring as a Bioisostere in Medicinal Chemistry. Drug Discov Today. 2017; 22: 1572–1581.
- Feng LS, Zheng MJ, Zhao F, Liu D. 1,2,3-Triazole Hybrids with Anti-HIV-1 Activity. Arch Pharm. 2021; 354: e2000163.
- Yan M, Xu L, Wang Y, Wan J, Liu T, Liu W. Opportunities and Challenges of Using Five‐membered Ring Compounds as Promising Antitubercular Agents. Drug Dev Res. 2020; 81: 402–418.
- Rani A, Singh G, Singh A, Maqbool U, Kaur G, Singh J. CuAAC-ensembled 1,2,3-Triazole-Linked Isosteres as Pharmacophores in Drug Discovery: Review. RSC Adv. 2020; 10: 5610–5635.
- Al-blewi FF, Almehmadi MA, Aouad MR, Bardaweel SK, Sahu PK, Messali M, et al. Design, synthesis, ADME prediction and pharmacological evaluation of novel benzimidazole-1,2,3-triazole-sulfonamide hybrids as antimicrobial and antiproliferative agents. Chem Cent J. 2018; 12: 110–123.
- Saeedi M, Mohammadi-Khanaposhtani M, Pourrabia P, Razzaghi N, Ghadimi R, Imanparast S, et al. Design and synthesis of novel quinazolinone-1,2,3-triazole hybrids as new anti-diabetic agents: In vitro α-glucosidase inhibition, kinetic, and docking study. Bioorg Chem. 2019; 83: 161–169.
- Chu XM, Wang C, Wang WL, Liang LL, Liu W, Gong KK, et al. Triazole derivatives and their antiplasmodial and antimalarial activities. Eur J Med Chem. 2019; 166: 206–223.
- Srinivas S, Neeraja P, Naveen K, Banothu V, Dubey PK, Mukkanti K. Synthesis, Chemotherapeutic Screening and Docking Studies of NSAID Inserted Peptide‐Triazole Hybrid Molecules. Chemistry Select. 2020; 5: 6786–6791.
- Bozorov K, Zhao J, Aisa HA. 1,2,3-Triazole-containing Hybrids as Leads in Medicinal Chemistry: A Recent Overview. Bioorg Med Chem. 2019; 27: 3511–3531.
- Nuzziz A, Massi A, Dondoni A. Model studies toward the synthesis of thymidine oligonucleotides with triazole inter nucleoside linkages via iterative Cu(I)-promoted azide-alkyne ligation. QSAR Comb Sci. 2007; 26: 1191- 1199.
- Luo G, Chen L, Conway CM, Denton R, Keavy D, Signor L, et al. Discovery of (5S,6S,9R)-5-amino-6-(2,3- difluorophenyl)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-9-yl-4-(2-oxo2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxylate (BMS927711): an oral calcitonin gene-related peptide (CGRP) antagonist in clinical trials for treating migraine. J Med Chem. 2012a; 55: 10644-10651.
- Wang Y, Li P, Liang X, Ye J. Base–Base Bifunctional Catalysis: A Practical Strategy for Asymmetric Michael Addition of Malonates to α,β-Unsaturated Aldehydes. Adv Synth Catal. 2008; 350: 1383-1389.
- Guo J, Wang X, Ni C, Wan X, Hu J. Nat Commun. SulfoxFluor-enabled deoxyazidation of alcohols with NaN3. Nature Communications. 2022; 13: 2752.
- Ma Y, Jiao X, Wang Z, Mu H, Sun K, Li X, et al. Organic Process Research & Development. 2022; 26: 1971-1977.
- Thotla K, Noolea VG, Kedikaa B, Krishna Reddy CH. Synthesis of 5-{[(1-Aryl-1H-1,2,3-triazol-4-yl)methyl]sulfanyl}-1-phenyl-1H-tetrazoles. Russian Journal of Organic Chemistry. 2020; 56: 1077–1081.
- Gurusamy Mariappan, Rajib Hazarika, Faruk Alam, Rashmi Karki, Uddhav Patangia, Shyamalendu Nath. Synthesis and biological evaluation of 2-substituted benzimidazole derivatives. Arabian Journal of Chemistry. 2015; 8: 715-719.
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007; 450: 712-716.
- Yuan H, Wang Z, Li L, Zhang H, Modi H, Horne D, et al. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood. 2012; 119: 1904–1914.
- Zhao X, Allison D, Condon B, Zhang F, Gheyi T, Zhang A, et al. The 2.5 Å crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J Med Chem. 2013; 56: 963-9.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Computational Chemistry. 2009; 16: 2785-91.
- Gandham SK, Kudale AA, Allaka TR, Chepuri K, Jha A. Novel tetrazolopyrrolidine–1,2,3–triazole analogues as potent anticancer agents: Design, synthesis and molecular docking studies. Mol Divers. 2023.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017; 7: 42717.
- Venkateswara Rao G, Allaka TR, Gandla MK, Veera Venkata Nanda PK, Pindi SR, Vaddi PRR, et al. Synthesis, antimicrobial activity and in silico studies of fluoroquinolones bearing 1,3,4–oxadiazolyl–triazole derivatives. J Heterocycl Chem. 2023; 60: 1666‒1683.
- Bakchi B, Dileep Krishna A, Sreecharan E, Jaya Ganesh VB, Niharika M, Maharshi S, et al. An overview on applications of SwissADME web tool in the design and development of anticancer, antitubercular and antimicrobial agents: A medicinal chemist’s perspective. Journal of Molecular Structure. 2022; 1259: 132712.
- Reddy AB, Avuthu VSR, Kishore PVVN, Allaka TR, Nagarajaiah H. New triazole based oxadiazolo/thiadiazolo–phthalazines as potent antimycobacterial agents: Design, synthesis, molecular modelling and in silico ADMET profiles. Chemistry Select. 2024; 9: e202304020.