
Special Article - Antisense Drug Research and Development
J Drug Discov Develop and Deliv. 2016; 3(1): 1019.
History and Properties of Morpholino Antisense Oligos
Summerton J*
Gene Tools, LLC, USA
*Corresponding author: Summerton J, Gene Tools, LLC, 1001 Summerton Way, Philomath, Oregon 97370, USA
Received: January 28, 2016; Accepted: April 04, 2016; Published: April 06, 2016
Abstract
This paper provides a very brief overview of the beginning of the antisense field, including the first antisense structural types, first patent, and first company in the field. The evolution of the Morpholino antisense structural type is then described in some detail, starting with Carbamate-DNA, then Carbamate- Morpholino, and finally the motivation for and development of the current Phosphorodiamidate-Morpholino structural type.
The current Morpholino structural type is then compared with the main competing antisense structural types: S-DNA, PNA, and siRNA. Finally, the challenges of in vivo delivery are briefly discussed, and the promise of new developments in this regard, as well as an exciting new application of Morpholinos which it is hoped will soon lead to safe, effective, and affordable cures for virtually all cancers - possibly by 2020.
Keywords: History of morpholinos; Morpholino versus siRNA
Introduction
Birth of the antisense therapeutics strategy
The antisense therapeutics strategy entails using a strand of genetic material, or a specially designed analog of genetic material, (the antisense drug) to very specifically bind and thereby block or destroy a complementary sequence of RNA or DNA (the sense target). In principle this antisense strategy offers the possibility of safe and effective drugs for a host of currently un-treatable or poorly-treatable diseases, such as viral diseases, cancers, some genetic defects, and many other diseases and conditions. However, in practice there were a number of daunting technical challenges that had to be surmounted in order to go from that simple antisense principle to safe, effective, and affordable antisense drugs based on that principle.
From 1967 through the end of the 1970s at least four groups independently worked on this antisense strategy, apparently with each group being unaware of the other groups’ activities. These groups included: Belikova, Zarytova & Grineva [1], three women scientists at the Academy of Science of the USSR in Novosibirsk, Siberia; Miller and Ts’o [2,3] at Johns Hopkins Univ.; Summerton (myself) & Bartlett [4-6] at Berkeley, Zamecnik & Stephenson [7,8] at Harvard. In 1978 the first patent on such antisense agents issued to me & Bartlett, and was assigned to Nat Inst of Health (US Patent 4,123,610). In 1980 I founded the first antisense company focused on developing and commercializing antisense drugs. That company was “ANTIVIRALS, Inc. (AVI)”, subsequently renamed “AVI Biopharma”, and more recently renamed “Sarepta Therapeutics”. (In 1997 I left AVI to found GENE TOOLS, LLC, focused on providing custom-sequence Morpholinos to the research community).
In the early 1980s my company, AVI, and various academic research groups investigated a substantial number of possible antisense structural types, and by 1984 two antisense structural types stood out as showing the most promise for future therapeutic applications. One was Methylphosphonate-DNA (Figure 1) developed at Johns Hopkins by Miller and Ts’o [3]. The other was Carbamate- DNA (Figure 1) developed at AVI by me, with much chemical advice from Dwight Weller in the Chemistry Department at Oregon State Univ [9]. A comparison of the advantages and limitations of these two leading structural types is below (Table 1).
![]()
Properties
DNA
Methylphosphonate-DNA
Carbamate-DNA
Resistance to degradation
poor
excellent
excellent
Aqueous solubility
good
poor
poor
Binding affinity to RNA
high
low
high
Cost of starting materials
expensive
expensive
expensive
Easy to assemble
no
no
Yes
Table 1: Properties of DNA analogs.
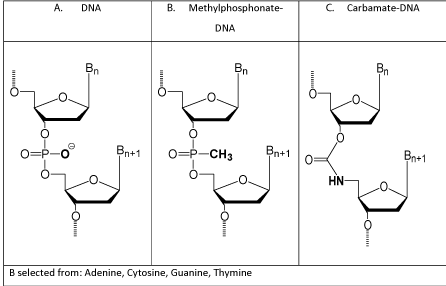
Figure 1: Early antisense structural types.
By the mid-1980s a great deal of interest had developed in the antisense therapeutics strategy, with substantial NIH (US National Institutes of Health) grant funding being provided to academic research groups. Most of the major pharmaceutical companies were also beginning to show a serious interest in this therapeutics strategy. There was also talk of new antisense biotechnology companies being organized - though actual startup of the new antisense companies did not occur until 1987, 1988, and 1989, most with venture capital backing.
Birth of morpholino antisense oligos
After working extensively with our Carbamate-DNA structural type, and closely monitoring the work of Miller and Ts’o, and other groups studying the Methylphosphonate-DNA structural type, I concluded that neither type was likely to ever be fully suitable for the broad range of medical applications I believed the antisense therapeutics strategy promised. Therefore, in the Fall of 1984 I set out to devise an alternative antisense structural type that would better meet the demanding design challenges for antisense therapeutic applications: (i) resistance to enzymatic degradation; (ii) high binding affinity for complementary RNA; (iii) excellent specificity for targeted RNA sequence; (iv) good solubility in aqueous solution; (v) predictable targeting; (vi) freedom from off-target effects; and, (vii) affordable production costs.
Of particular note, I estimated that the then-available DNA-based antisense structural types, when scaled up to doses likely to be needed for patients, could cost from hundreds of thousands to millions of dollars per patient. To help reduce such exorbitant costs I set out to devise an antisense structural type that was not derived from the very expensive DNA subunits (deoxyribonucleosides) used in the antisense structural types developed to that time, but instead would be derived from the 30-fold-cheaper RNA subunits (ribonucleosides). My goal was also to devise some way to cheaply introduce an amine into the backbone structure - on the premise that could substantially reduce costs and raise efficiency of assembling the antisense drug by removing the need for: 1) ultra-dry reagents (a significant cost factor); 2) expensive catalysts in the assembly steps; and, 3) a coupling reagent that required an oxidation step during or at the end of oligo assembly (“oligo” denotes a short chain of subunits).
After cogitating for weeks on how such changes might be accomplished, and building many prospective alternative structures with my CPK (Corey, Pauling, Koltun) molecular models, finally on 1 January 1985 I came up with a rather radical departure from the natural, and the slightly modified, DNA and RNA structures which had been developed to that time. That radical structural departure entailed replacing the 5-membered sugar backbone moieties of RNA and DNA with 6-membered “morpholino” backbone moieties. To assure resistance to enzymatic degradation, the negatively charged phosphate inter subunit linkages of DNA and RNA were replaced with uncharged carbamate inter subunit linkages. This new structural type, shown in (Figure 2), satisfied my objectives of: (i) use of far cheaper ribonucleosides starting materials; (ii) replacing a hard-tocouple hydroxyl with an easy-to-couple amine; (iii) a relatively simple and inexpensive synthetic route to the Morpholino subunit structures; and, (iv) a very simple and highly efficient oligomer assembly method (relative to DNA and RNA assembly methods).
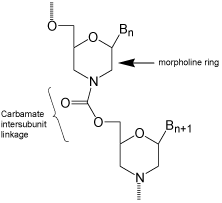
Figure 2: Carbamate-morpholino.
The subunit synthesis steps are shown in (Figure 3). It is noteworthy that steps 1, 2, and 3 of (Figure 3) constitute a simple onepot synthesis with no intervening workups. The assembly of subunits of (Figure 3) into antisense oligos entails a simple two-step coupling cycle for adding each subunit to the growing oligo, shown in (Figure3).
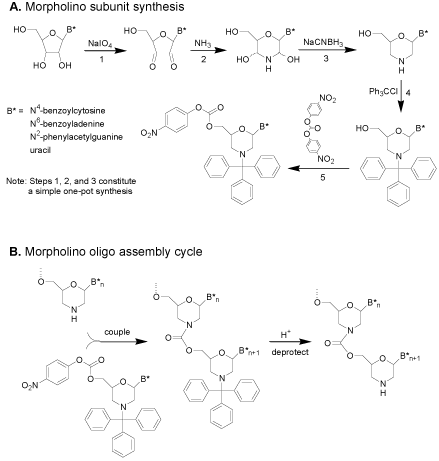
Figure 3: Initial carbamate-morpholino synthesis.
This newly-envisioned Morpholino structural type looked promising from a synthetic standpoint, and my CPK molecular modeling suggested that the new antisense structural type should allow good Watson/Crick pairing to a complementary genetic sequence. Therefore, later that day (New Years Day) I called Dr. Dwight Weller of the OSU Chemistry Dept., who often advised me on organic synthesis matters, and he assured me that my proposed synthesis route looked quite feasible - and he suggested one small upgrade in the choice of a reducing agent.
I also called Dr. Donald Johnson, Director of New Technology Research at DuPont, to apprise him of a likely change in our program to develop non-ionic antisense agents. Dr. Johnson invented DuPont’s highly successful Automated Clinical Analyzer, and he directed its development. He was the person who initially approached ANTIVIRALS, Inc. regarding an offer from DuPont to provide modest research funding in return for the rights to use non-ionic antisense structural types developed at AVI for diagnostic applications. (Both Dr. Johnson and I were aware that non-ionic probes can provide huge advantages in a probe diagnostic system.) When I called, he assured me that if my newly-devised non-ionic Morpholino structural type succeeded then DuPont would be pleased to adjust our funding agreement to accommodate that new structural type.
Soon thereafter I also contacted the National Cancer Institute and the National Institute of Allergy and Infectious Diseases.
Infectious Diseases, which were providing ANTIVIRALS Inc. with modest ($50,000) Phase 1 SBIR (Small Business Innovation Research) grants. I informed the respective grants managers that I had a new antisense structural type and was requesting their approval to shift my grant funds to development of this new more promising Morpholino structural type. My requests were denied and so I relinquished the unspent portions of those grants. I gave up those grants because I had no intention of continuing to spend time and funds on the Carbamate-DNA structural type, which by that time appeared to me to be a dead-end structural type that was unlikely to adequately fulfill the tremendous promise of antisense therapeutics.
Developing this new Carbamate-Morpholino antisense structural type took more than a year. It entailed converting a cytosine ribonucleoside to its cytosine morpholino derivative, as shown in (Figure 3), assembling (via carbamate intersubunit linkages) a short chain of such morpholino subunits, as shown in (Figure 3), and testing that oligo for binding to its complementary DNA oligo as a function of temperature. That temperature study provided a precise measure of the binding affinity of the Morpholino oligo for its complementary DNA oligo. Note that while it was clear our ultimate targets would be RNA, our initial binding studies were typically carried out with a DNA complement instead of an RNA complement. This was because in the mid-1980s defined DNA oligos were far cheaper to buy than the corresponding RNA oligos, and in those early days at AVI finances were extremely limited.
As hoped, that Morpholino oligo showed an excellent binding affinity for its DNA complement.
With that very positive result in hand, the next step was to synthesize the other three genetic letters (A, G, and U) in the morpholino series, and then assemble them into a longer oligo with a specific sequence of genetic letters, and finally test that Carbamate- Morpholino oligo for binding to its complementary DNA as a function of temperature. Again, this more complex Morpholino oligo showed exceptionally good binding to its complementary DNA, and no binding to a non-complementary DNA.
At that point it appeared we had a winning antisense structural type which could be on the order of 50-fold less expensive to make than the competing Methylphosphonate-DNA structural type, and compared to the Methylphosphonate-DNA, the Carbamate- Morpholino had a much higher binding affinity for its complementary DNA-allowing its use at much lower concentrations, thereby further reducing the cost of antisense drugs.
However, since virtually all of the targets for antisense therapeutics would be single-stranded RNAs (RNA transcripts) we next purchased a very expensive complementary RNA and assessed that same Carbamate-Morpholino oligo’s binding to that RNA strand as a function of temperature [10].
Backbone problem
We were appalled by the result of the Carbamate-Morpholino/ RNA binding assay. Our Carbamate-Morpholino oligo did not bind to its complementary RNA. After recovering from the huge shock of that result, we spent more than a week trying to understand why the Morpholino antisense oligo could show such excellent binding to DNA, but virtually no binding to the same sequence of genetic letters in the very similar RNA.
Finally a possible explanation began to emerge from my CPK molecular modeling comparisons of the Morpholino/DNA duplex (which can adopt a B conformation) and the Morpholino/RNA duplex (which can only adopt the more constrained A conformation). From the molecular models it appeared that the carbamate inter subunit linkages would be in their energetically favored planar conformation in the Morpholino/DNA duplex. In contrast, it appeared that in the Morpholino/RNA duplex the carbamate linkages would be forced to exist in their energetically unfavorable non-planar conformation. Subsequently, a detailed computer modeling study by a collaborating group at DuPont further supported our postulate for why the Carbamate-Morpholino oligo failed to bind its complementary RNA.
Once we had a likely explanation for why the Carbamate- Morpholino oligo failed to bind RNA, we postulated that this failureto- bind problem could be fixed by going to a more flexible inter subunit linkage type. Subsequently, both our and DuPont’s molecular modeling studies suggested that Morpholino oligos with more flexible inter subunit linkages should indeed allow good binding to both DNA and the all-important RNA.
Because the fundamentals of the Morpholino antisense structural type appeared so promising, rather than abandoning this “failed” antisense structural type I decided instead to focus our efforts on finding a suitable replacement linkage that would provide the increased flexibility predicted to allow good binding to complementary RNA. Finding a suitably flexible linkage, which would also be easy to form and adequately stable, turned out to require substantially more time and effort than expected (two years). However, by 1989 that quest was a smashing success - giving (after many failures) our current very effective antisense structural type shown in (Figure 4).
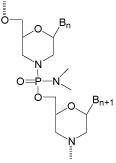
Figure 4: Ultimate phosphorodiamidate-morpholino.
Once we had this new Phosphorodiamidate-Morpholino structural type, we next spent substantial time refining the methods for the subunit syntheses, as well as the oligo assembly methods. Then we moved on to biophysical and biochemical assessments (solubility assays, binding affinity assessments, testing for chemical stability and stability against a range of degradative enzymes). In every case the new structural type came through with flying colors.
We then moved on to biological assessments. Initially, we used a cell-free reticulocyte lysate translation system to assess the ability of the Morpholino oligos to block translation of selected messenger RNAs (the two mRNAs coding for the alpha and the beta subunits of hemoglobin). In that system we assessed efficacy as a function of: oligo length, oligo concentration, the fraction of G+C genetic letters in the oligo, and the impact of experimentally-determined stem/ loop secondary structures in the target RNA sequences on the rate and extent of oligo/target pairing. We also did detailed studies of the efficiency of target inactivation as a function of the position of the target site along the length of the mRNA. We further used that two-mRNA system to rigorously assess specificity by targeting one of the two mRNAs and then precisely comparing the relative ratios of the two translated protein products (the alpha and beta hemoglobin subunits which are easily separated by gel electrophoresis). Most of those biophysical, biochemical, and biological studies also included direct comparisons between our Morpholino oligos and competing antisense structural types - including the then new Phosphorothioate- DNA structural type (often referred to as S-DNA).
We then moved on to biological assessments. Initially, we used a cell-free reticulocyte lysate translation system to assess the ability of the Morpholino oligos to block translation of selected messenger RNAs (the two mRNAs coding for the alpha and the beta subunits of hemoglobin). In that system we assessed efficacy as a function of: oligo length, oligo concentration, the fraction of G+C genetic letters in the oligo, and the impact of experimentally-determined stem/ loop secondary structures in the target RNA sequences on the rate and extent of oligo/target pairing. We also did detailed studies of the efficiency of target inactivation as a function of the position of the target site along the length of the mRNA. We further used that two-mRNA system to rigorously assess specificity by targeting one of the two mRNAs and then precisely comparing the relative ratios of the two translated protein products (the alpha and beta hemoglobin subunits which are easily separated by gel electrophoresis). Most of those biophysical, biochemical, and biological studies also included direct comparisons between our Morpholino oligos and competing antisense structural types - including the then new Phosphorothioate- DNA structural type (often referred to as S-DNA).
By the mid-1990s we had a thorough understanding of the functioning of our new Morpholino antisense type at the molecular level, and so we began rigorous studies at the next level of complexity, that being in cultured cells. Our test system entailed delivering plasmids (small engineered circles of DNA) into several lines of cultured human cells. Those plasmids contained genetic sequences which could be induced to produce the enzyme (luciferase) which fireflies use to produce light. When an inducer compound was added to those cells it would cause the plasmid to generate a messenger RNA which would be used to produce luciferase which generated light which we measured in a luminometer (an extremely sensitive light meter). However, when a suitably targeted antisense oligo was also delivered into such cells, that antisense oligo could block the production (translation in the cytosol of the cells) of that light-producing enzyme - resulting in much less light being produced. Using that complicated, but quite reliable and quantitative translation-blocking test system, we found that Morpholinos had excellent efficacy and specificity in these cultured human cells [11]. A few years later we and many others in the antisense field switched over to a much simpler and even more robust test system which was developed by Ryszard Kole at Univ. of North Carolina. Kole’s splice-correction test system instead measured antisense activity on a targeted RNA in the nucleus of cells [12].
In these head-to-head biophysical, biochemical, and biological comparisons our Morpholinos outshone all the other antisense structural types with respect to a composite of key properties [13-16]. Of particular note, our Morpholinos were shown to be more effective and far more specific than S-DNAs, the most popular of the other antisense types during the 1990s [17].
The most challenging of all antisense applications is in studies of developing embryos where intricate cascades of gene activations and deactivations are precisely controlled with respect to both time and position in the rapidly maturing embryo. For studies in such a complicated system it is essential that the antisense oligos: (i) provide exquisite specificity for their targeted RNA; (b) achieve thorough inactivation of their targeted RNA; (c) be largely free of off-target effects; and, (d) remain stable in biological systems throughout multiple days of embryonic development. Because Morpholinos provide this unmatched combination of compelling advantages, they are the only gene-modulating tools which can routinely provide reliable results in developing embryos (particularly frogs and zebrafish). For this reason, since the year 2000 Morpholinos have become the essential tools for most researchers in developmental biology, and the use of Morpholinos has revolutionized that very demanding field of research [18-20]. Scientists using Morpholinos have published over 7,500 research papers (searchable at: pubs.genetools. com) wherein these precision tools played a key role in the reported experiments, and over half of those publications came from developmental biology researchers.
Competition
At ANTIVIRALS Inc. from the beginning I believed the best way to successfully achieve the promise of antisense therapeutics was to rigorously test our new antisense structural types in systems of progressively increasing complexity. This allows maximal rigor in the simplest systems, and then progressive tradeoffs between rigor and complexity as one moves from biophysical studies, to biochemical studies, to cell-free biological studies, to studies in cultured cells, to studies in small animals, and finally to humans. This systematic approach allows one to build a solid foundation and then to rigorously build a solid edifice of experimental results on that foundation - hopefully leading to the final goal of safe, effective, and affordable treatments for a host of currently un-treatable and poorly-treatable diseases (such as cancers).
However, such a systematic approach generally does not sit well with business types, venture capitalists, and investors. That systematic approach also runs a real risk of one’s development program getting scooped, or at least of having the appearance of being scooped, by competing groups who elect to skip the early time-consuming levels of testing, and instead jump directly to a complexity level more likely to impress investors. However, there is a substantial tradeoff to jumping immediately to a complex test system - that being it creates a serious risk of moving forward with an antisense structural type which suffers from severe fundamental flaws that can easily be overlooked in more complex, but much less rigorous, test systems. Figure 5 illustrates the three main antisense structural types which have been in competition with Morpholinos since the early 1990s.
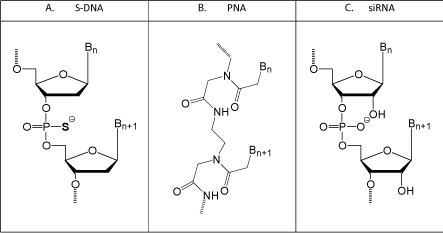
Figure 1: Newer (but not as good) antisense types.
S-DNAs are a good example of the risks of a rush to do initial testing in complex systems without first confirming that the structural type is free of severe fundamental and uncorrectable flaws. While Fritz Eckstein and coworkers at the Max-Plank Inst. in Germany studied thiophosphate linkages in DNA in the 1960s, 1970s, and 1980s, it was not until 1987 that scientists (at the National Cancer Institute and the US Food and Drug Administration) first prepared phosphorothioatelinked DNA antisense oligos (S-DNAs, shown in (Figure 5), and then they apparently jumped directly to targeting HIV (the AIDS virus) in cultured cells [21]. This was followed a year later by another group’s report of also using S-DNAs for targeting that same virus in cultured cells [22]. Even in those first two papers on use of S-DNA antisense oligos there should have been very serious concerns raised by the authors of these papers about this structural type because the control S-DNA oligos (which were designed to not show activity) showed activities almost as good as, and in some cases much better than the designed S-DNA antisense oligos. In light of the results with the controls, the authors should have begun a series of rigorous studies at the biophysical, biochemical, and/or biological levels to determine why the S-DNA controls showed such high activity relative to the actual antisense oligos.
Instead, from the first report in 1987 over the next few years use of that structural type grew to the point that by the 1990s the S-DNA antisense structural type had taken the antisense field by storm and continued to dominate the antisense field until about 2005. In particular, a majority of the funds going into the antisense field after about 1990 were focused on animal studies and clinical trials on that still-very-questionable S-DNA antisense structural type. What the S-DNAs had going for them was: (i) relative to the early Methylphosphonate-DNA type, S-DNAs achieved high levels of target inactivation (but not as high as our new Morpholinos); and, (ii) relative to DNA, S-DNAs had greater resistance to enzymatic degradation (but while S-DNAs are more stable than DNA, the stability of S-DNAs is still woefully inadequate for longer-term applications). It also helped that the business community, and many scientists, were much enamored with S-DNAs because: (iii) they looked almost identical to natural DNA (compare Figure 1 and Figure 5); (iv) they exploited a natural enzyme (RNase H) to achieve their high efficacies; (v) they were developed at NIH, the nation’s premier medical research facility; and, (vi) they could be made in large quantities by the methods previously worked out for DNA, with just the oxidation step changed. Based on that already-developed synthesis capability, many in the business community were led (by several biotech companies) to expect a whole host of multi-billion dollar S-DNA drugs for treating a multitude of severe diseases - with the further expectation that such S-DNA therapeutics could be rapidly developed, then rapidly carried through clinical trials, and thereafter marketed in record time - bringing great wealth to investors and great benefits to patients.
I estimate that well over a billion dollars of grant funding and drug development funds were wasted on S-DNAs between about 1990 and 2005 - before it finally became clear to most scientists that S-DNAs have severe fundamental limitations which preclude their use as safe and effective therapeutics. The fundamental problem with “S”-DNAs is the “S” (sulfur atom) on each intersubunit linkage. That pendant anionic sulfur has now been documented to bind strongly to many different proteins outside of cells, on cell surfaces, and within cells, and this strong binding leads to the host of off-target effects for which S-DNAs are notorious. Further, the widely touted increase in efficacy, which is due to RNase H enzyme cleaving the RNA targets bound by S-DNA oligos, turns out to be the culprit responsible for the very poor specificities which plague S-DNA oligos. The problem is that when an S-DNA oligo transiently pairs with a partially-complementary nontarget RNA, resultant duplexes as short as 5 base-pairs can be cleared by R Nase H. As a consequence, virtually every S-DNA antisense oligo is expected to cause inadvertent destruction of thousands (estimated about 3,500 in a human cell [15,16]) of non-targeted RNA species in a typical human cell. Since the pendant sulfurs in an S-DNA antisense oligo are essential to its high efficacy and increased resistance to degradation in biological systems, those sulfurs constitute a severe and uncorrectable flaw in the S-DNA structural type. Even when mixed structural types are used, comprising a central S-DNA segment to exploit R Nase H cleavage of the target RNA, with segments of a higher-binding-affinity structural type at each end, one is still left with limited efficacy, poor specificity, excessive off-target effects, and inadequate stability in biological systems - resulting in very suboptimal safety and efficacy for any antisense structural type which contains significant S-DNA content.
PNA (Peptide Nucleic Acid, (Figure 5)).
Peptide Nucleic Acids (PNAs) were developed by Nielsen and Egholm in the early 1990s, and use of PNAs as antisense oligos was first reported in 1993 [23]. In sharp contrast to the case for S-DNAs, PNAs were rigorously tested in systems of progressively increasing complexity and so their capabilities and limitations were well defined from their earliest days. The resulting detailed information on their properties informed development of a wide variety of novel applications unmatched even by Morpholinos [24,25]. In particular, PNAs’exceptionally high binding affinity for DNA and RNA, coupled with their high conformational flexibility, lack of ionic charge, and resistance to degradation in biological systems, allows their advantageous use for detection of single-base mutations, and allows their invasion of double-stranded DNA under low salt conditions. Their special combination of properties also allows their use for targeting duplex DNA sequences having a run of purines in one strand - to which the PNA oligo can strongly adhere via. Hoogsteen or reverse Hoogsteen binding to major groove sites of the duplex DNA.
However, for the special case of antisense therapeutic applications in complex systems (in animals, including humans) the composite of properties of Morpholinos make the Morpholino structural type preferred over PNAs as antisense drugs. The relevant properties of PNAs and Morpholinos have been thoroughly compared in Chapter 6 of the book: “Peptide Nucleic Acids, Morpholinos and Related Antisense Biomolecules”, published in 2006 [15].
As described on pages 95 and 96 of ref [15], PNAs have significantly greater backbone flexibility than Morpholinos. While this is a distinct advantage for PNAs in some applications, in the context of therapeutic applications that greater conformational flexibility substantially increases internal self-pairing, which markedly limits their targeting success rate, relative to the exceptionally high targeting success rate for the conformationally more rigid Morpholinos.
A key property of PNAs is their exceptionally high affinity for complementary RNA - and this high affinity underlies a number of the unmatched special applications of PNAs. However, this same very high affinity for RNA substantially reduces the length of antisense oligo required to achieve good target blocking activity - which substantially reduces their specificity in complex biological systems. This counter-intuitive consequence of high binding affinity is explained and demonstrated experimentally on pages 98 through 104, and particularly in Figure 11, in reference [15]. Thus, overall PNAs are appreciably less effective than Morpholinos for therapeutic applications. This is borne out by PNAs’ inability to provide reliable antisense results in developing embryos.
siRNA (short interfering RNA, (Figure 5))
In 1986 Ecker, Davis, and Davis reported what was ultimately learned to be in essence a natural antisense system in plants. Further study in the 1990s showed that such natural antisense systems were also widely present in animals all the way up through humans. By 2001 duplexes of 21-nucleotide RNAs (siRNAs) were successfully used by Tuschl and coworkers as natural antisense oligos in a variety of human cell lines [26]. Since that time siRNAs have been widely used for gene knockdown in cancers, against viral targets, and against many other targets [27].
Not surprisingly, siRNAs have been extensively tested in zebrafish embryos. A 2005 paper by Tuschl and coworkers [28] briefly summarizes their findings:
“The unspecific effects of siRNA on embryonic development seen in this and other studies indicate that siRNAs in the zebrafish have an unspecific effect. Thus, currently RNAi is not a useful technique for studying gene function in zebrafish embryos and the morpholino technique where modified oligonucleotides block translation of the corresponding mRNA is clearly preferable.”
Based on published reports, it appears that, relative to Morpholinos, the siRNAs suffer the following limitations: (i) substantially poorer sequence specificity (because their 11-base seed sequence recognizes too little sequence information in the targeted RNA transcripts); (ii) limited stability in biological systems (because siRNAs are degraded by nucleases both outside and inside of cells); (iii) substantially poorer targeting predictability (because high complementarity to the targeted RNA transcript leads to cleavage of the targeted RNA, while lesser complementarity can lead to substantial, but unpredictable, translation inhibition of a multiplicity of partially-complementary non-targeted RNA transcripts) and (iv) inability to modify splicing (because the RISC complex is located in the cytosol).
Delivery
Delivery of antisense oligos of all types into the cytosol of a broad range of different cell types in living animals (in vivo) has proven to be one of the most difficult challenges for antisense therapeutics. Many antisense therapeutics developers have elected to use polycationic delivery components because such components are relatively effective for delivering a broad range of substances into the cytosol of cells. However, delivery with poly-cationic components generally causes significant toxicity, both in cultured cells and in vivo.
Since the 1990s my companies (initially ANTIVIRALS, Inc, and since 1997, GENE TOOLS, LLC) have developed 5 antisense delivery systems, with each being an improvement over the last. However, to date none has been truly adequate for safe, efficient, and affordable delivery of antisense therapeutics in vivo.
However, three years ago we began development of a novel 4-component delivery system which appears truly adequate for safe, efficient, and affordable delivery of antisense therapeutics into cultured cells (with medium containing up to 10% serum). We plan to launch this into the research market in May of 2016.
Regrettably, the 4th component of that delivery system does not yet work well in the presence of the high serum concentration present in the blood of mammals. Accordingly, we are currently working to modify that 4th component to make it compatible with the high serum concentration it will face in vivo. Based on preliminary improvements achieved to date, we hope to achieve a successful modification of that 4th component before the end of 2016, and then soon thereafter launch the resulting 4-component in vivo delivery system which will allow safe, efficient, and affordable delivery of Morpholino drugs for treating a broad range of currently un-treatable or poorly-treatable diseases.
An exciting new application for morpholinos
In May of 2016 GENE TOOLS plans to make available to the oncology research market affordable delivery-enhanced precisiontargeted Morpholino antisense drugs specific for most of the estimated several thousand cancer-related messenger RNAs known in human cancers. These cancer-targeted Morpholino drugs will allow researchers in the oncology field to efficiently and decisively identify cancer-essential transcripts in a broad range of human cancers, where cancer-essential transcripts are defined as those transcripts, identified by sequencing a biopsy sample of a patient’s cancer, which: a) are not present in normal cells of adults (so can be targeted without damaging the patient); b) are present in a given patient’s cancer (transcribed from embryo-active genes normally deactivated in adults, but reactivated by mutation in at least most, but probably all cancers, or from oncogenic viruses); and, c) are essential to the viability of that cancer.
I postulate that a custom cocktail of such Morpholino drugs targeted against a number of those cancer-essential genes found by sequencing to be present in a given patient’s cancer, will provide a decisive (and affordable) cure for that patient’s cancer - without any damage to the patient. This “custom cocktail” strategy for destroying any cancer, including probably late-stage metastatic cancers, without harm to the patient, is described in another paper in this journal issue.
References
- Belikova A, Zarytova V, Grineva N. Synthesis of ribonucleosides and diribonucleoside phosphates containing 2-chloro-ethylamine and nitrogen mustard residues. Tetrahedron Letter. 1967; 37: 3557-3562.
- Miller P, Barrett J, Ts’o P. Synthesis of oligodeoxyribonucleotide ethyl phosphotriesters and their specific complex formation with transfer ribonucleic acid. Biochemistry. 1974; 13: 4887-4986.
- Smith C, Aurelian L, Reddy M, Miller P, Ts’o P. Antiviral effect of an oligo (nucleoside methyl phosphonate) complementary to the splice junction of herpes simplex virus type 1 immediate early pre-mRNA 4. Proc Natl Acad Sci USA. 1986; 83: 2787-2791.
- Summerton J. Intracellular inactivation of specific nucleotide sequences: a general approach to the treatment of viral diseases and viral-mediated cancers. J Theor Biol. 1979; 78: 77-99.
- Summerton J, Bartlett PA. Sequence-specific cross linking agents for nucleic acids. Use of 6-bromo-5,5-dimethoxyhexanohydrazide for cross linking cytidine to guanosine and cross linking RNA to complementary sequences of DNA. J Mol Biol. 1978; 122: 145-162.
- Summerton J. Sequence-specific crosslinking agents for nucleic acids: design and functional group testing. J Theor Biol. 1979; 78: 61-75.
- Zamecnik P, Stephenson M. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci USA. 1978; 75: 280-284.
- Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. 1978; 75: 285-288.
- Stirchak E, Summerton J, Weller D. Uncharged stereoregular nucleic acid analogues. Synthesis of a cytosine-containing oligomer with carbamate internucleoside linkages. J Org Chem. 1987; 52: 4202-4206.
- Stirchak E, Summerton J, Weller D. Uncharged stereoregular nucleic acid analogues. 2. Morpholino nucleoside oligomers with carbamate internucleoside linkages. Nucleic Acids Res. 1989; 17: 6129-6141.
- Partridge M, Vincent A, Matthews P, Puma J, Stein D, Summerton J. A simple method for delivering morpholino antisense oligos into the cytoplasm of cells. Antisense Nucleic Acid Drug Dev. 1996; 6: 169-175.
- Kang S, Cho M, Kole R. Up-regulation of luciferase gene expression with antisense oligonucleotides: Implications and applications in functional assay development. Biochemistry. 1998; 37: 6235-6239.
- Summerton J, Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997; 7: 187-195.
- Summerton J. Morpholino antisense oligomers: the case for an R Nase H-independent structural type. Biochim Biophys Acta. 1999; 1489: 141-158.
- Summerton J. Morpholinos and PNAs compared. In: Peptide Nucleic Acids, Morpholinos and Related Antisense Biomolecules. 2006; 89-113.
- Summerton JE. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr Top Med Chem. 2007; 7: 651-660.
- Summerton J, Stein D, Huang S, Matthews P, Weller D, Partridge M. Morpholino and phosphorothioate antisense oligomers compared in cell-free and in-cell systems. Antisense and Nucleic Acid Drug Dev. 1997; 7: 63-70.
- Ekker SC. Morphants: a new systematic vertebrate functional genomics approach. Yeast. 2000; 17: 302-306.
- Heasman J. Morpholino oligos: making sense of antisense? See comment in PubMed Commons below Dev Biol. 2002; 243: 209-214.
- Special Issue: Morpholino Gene Knockdowns - all 27 research reports in the July issue of the journal: Genesis. 2001; 30: 89-200.
- Matsukura M, Shinozuka K, Zon G, Mitsuya H, Reitz M, Cohen JS, et al. Phosphorothioate analogs of oligodeoxynucleotides: Inhibitors of replication and cytopathic effects of human immunodeficiency virus. Proc Natl Acad Sci USA. 1987; 84: 7706-7710.
- Agrawal S, Goodchild J, Civeira MP, Thornton AH, Sarin PS, Zamecnik PC. Oligodeoxynucleoside phosphoramidates and phosphorothioates as inhibitors of human immunodeficiency virus. Proc Natl Acad Sci USA. 1988; 85: 7079-7083.
- Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, et al. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. 1993; 365: 566-568.
- Nielsen P, Egholm M. Norfolk Peptide Nucleic Acids Protocols and Applications. Horizon Scientific Press. 1999.
- Nielsen P. The many faces of PNA. In: Peptide Nucleic Acids, Morpholinos and related antisense biomolecules. 2006; 3-17.
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001; 411: 494-498.
- Scherer L, Rossi J. Recent applications of RNA Interference (RNAi) in mammalian systems. In: Peptide Nucleic Acids, Morpholinos and related antisense biomolecules. 2006; 133-147.
- Gruber J, Manninga H, Tuschl T, Osborn M, Weber K. Specific RNAi mediated gene knockdown in zebrafish cell lines. RNA Biol. 2005; 2: 101-105.