
Research Article
Austin J Endocrinol Diabetes. 2014;1(1): 1002.
The Ca2+/calmodulin-dependent protein kinase IIa (Thr286Asp) transgenic mice: a novel mouse model of severe insulin-dependent diabetes
Hikari Suzuki1, Shin Takasawa2*, Isao Usui3, Yoko Ishii4, Ichiro Kato5, Hiroshi Okamoto6, Masashi Kobayashi1, Masakiyo Sasahara4 and Kazuyuki Tobe3
1Social Insurance Takaoka Hospital, 8–5 Fushikikofumoto–machi, Takaoka, Toyama, 933–0115 Japan
2Department of Biochemistry, Nara Medical University, 840 Shijo–cho, Kashihara, Nara, 634–8521 Japan
3First Department of Internal Medicine, University of Toyama, 2630 Sugitani, Toyama, 930–0194Japan
4Department of Pathology, University of Toyama, 2630 Sugitani, Toyama, 930–0194Japan
5Tonami Sunshine Hospitals, 575 Takanosu, Tonami, Toyama, 939–1335 Japan
6Tohoku University, 2–1, Seiryo–machi, Aoba–ku, Sendai, Miyagi, 980–8575 Japan
*Corresponding author: Shin Takasawa, Professor and Chairman, Nara Medical University, 840 Shijo-cho, Kashihara, Nara, 634-8521 Japan
Received: January 02, 2014; Accepted: January 17, 2014; Published: January 20, 2014;
Abstract
Diabetes mellitus is the leading cause of blindness and end–stage renal disease. To understand the pathogenesis of diabetic complications, suitable animal models for this disease have been needed. The activation ofCa2+⁄ calmodulin–dependent protein kinase II (CaMKII) in pancreatic β–cells has been thought to play a central role in Ca2+–mediated insulin secretion. We generated transgenic mice over expressing the constitutively active–type CaMKIIα (Thr286Asp) in β–cells, which showed very high plasma glucose levels and exhibited the features of diabetic nephropathy and retinopathy. In cDNA microarray analysis osteopontin mRNA increased in CaMKIIα transgenic mice. In quantitative real–time RT–PCR analyses, not only M1 macrophage marker genes but also M2 macrophage marker genes were over expressed in renal cortex of CaMKIIα transgenic mice. The mice were crossed with conditional knockout mice in which platelet–derived growth factor receptor–β gene (Pdgfr–β) was deleted postnatal. The increased oxidative stress in the kidneys of the CaMKII α transgenic mice, which was shown by the increased urinary 8–hydroxydeoxyguanosine excretion and the increased expression of NAD (P) H oxidase 4, was decreased by Pdgfr–β deletion. The CaMKIIα (Thr286Asp) transgenic mice will be valuable as a novel model of severe insulin–dependent diabetes accompanied by an early progression of diabetic micro vascular complications.
Abbreviations
ATP: Adenosine triphosphate; cADPR: Cyclic adenosine diphosphate–ribose; CaM: Calmodulin; CaMKII:Ca2+⁄calmodulindependent protein kinase II; CCR2: CCmotif receptor 2; CHI3l3: Chitinase 3–like 3; DM: Diabetes mellitus; ERG: Electroretinography; IL–1β: ?nterleukin–1β; IP3 : Inositol 1,4,5–trisphosphate; MCP–1:Monocyte chemotactic protein–1; MGL2: Macrophage galactose N–acetyl–galactosamine specific lectin 2; MRC1: Mannose receptor C–type1; NAD+: Nicotinamide adenine dinucleotide; NOS2: Nitric oxide synthase 2; NOX4: NAD(P)H oxidase 4; 8–OHdG: 8–hydroxydeoxyguanosine; PDGF: Platelet–derived growth factor; PDGFRs: Platelet–derived growth factor receptors; RyR: Ryanodinereceptor; TG: Transgenic; TNFα: ?umor necrosis factor α; WT: Wild type.
Introduction and Background
Diabetes mellitus (DM) is a disease characterized by hyperglycemia and is caused by absolute or relative insulin deficiency, sometimes associated with insulin resistance [1]. As a consequence of its micro vascular pathology, DM is the leading cause of blindness, end–stage renal disease, and a variety of neuropathies [2]. Approximately 30% of type 1 DM patients suffered from diabetic nephropathy eventually undergo renal dialysis or transplantation [3]. Nephropathy is thus alife–threatening complication of DM and is the leading cause of endstage renal disease in developed countries. The features of diabetic nephropathy include persistent albuminuria, a progressive decline in renal function, and histopathologically mesangial expansion followed by glomerulosclerosis [4]. However, the molecular mechanisms leading to end–stage renal disease in DM have not been fully understood.
Analysis and Interpretation
Mechanism of Insulin Secretion
Cyclic ADP–Ribose in Insulin Secretion
Mobilization ofCa2+ from intracellular stores in the endoplasmic reticulum is required for insulin secretion from pancreatic β–cells. Inositol 1,4,5–trisphosphate (IP3) is thought to be a second messenger for intracellular calcium mobilization, while in islet microsomes cyclic adenosine diphosphate–ribose (cADPR) induces mobilization ofCa2+. In the process of glucose metabolism, adenosine triphosphate (ATP) is generated. ATP induces cADPR formation from nicotinamide adenine dinucleotide (NAD+)by inhibiting the cADPR hydrolase activity of CD38.CD38 has enzymic activites of both cADPR synthesis from NAD+(ADP–ribosyl cyclase activity) and cADPR hydrolysis to produce ADP–ribose (cADPR hydrolase activity) [5]. cADPR functions as a second messenger forCa2+ mobilization fromendoplasmic reticulum for glucose–induced insulin secretion from pancreatic β–cells [6].
cADPR Requires Calmodulin–Dependent Protein Kinase II For IntracellularCa2+ Mobilization
In sea urchin eggs, calmodulin (CaM) directly interacts with the ryanodine receptor (RyR) to enhance the cADPR–mediatedCa2+ release [7]. In rat islets, CaM sensitized and activated the cADPRmediatedCa2+ release from islet microsomes. It is reported that cADPR–mediatedCa2+ mobilization for insulin secretion is achieved by the activatedCa2+/calmodulin–dependent protein kinaseII (CaMKII) not by the direct interaction of CaM andCa2+ release [8].
Glucose stimulation to pancreatic β cell induces cADPR accumulation in islets by inhibiting cADPR hydrolase activity of CD38 by ATP. Glucose stimulation also activates CaMKII, and the activated kinase phosphorylates RyR to sensitize theCa2+ channel for the cADPR signal.As shown in Figure 1, cADPR acts as a second messenger for intracellularCa2+ mobilization via RyR, and inducesinsulin secretion [8–10]. Thus, CaMKII is suggested to be essential kinase for glucose–induced insulin secretion.

Figure 1: Possible involvement of CaMKII in cADPR mediated intracellular Ca2+ mobilization (adopted from [8]). cADPR binds to FK506 binding protein 12.6 (FKBP12.6) to release Ca2+ dissociating FKBP12.6 from RyR. CaMKII phosphorylates RyR to sensitize and activate the Ca2+ channel (Pi, phosphorylation of RyR by CaMKII). Ca2+, released from intracellular stores and/or supplied from extracellular sources, further activates CaMKII and amplifies the process. In this way, Ca2+–induced Ca2+ release can be explained. Increased Ca2+ activates many physiological⁄pathological events such as glucose-induced insulin secretion and cell death.
A Novel Mouse Model of Diabetic Nephropathy
Mouse Model of Diabetic Nephropathy
Spontaneously diabetic animals such as no obese diabetic mice developed only limited lesions of diabetic nephropathy such as mild mesangial sclerosis [11]. The same was the case with chemically induced diabetic rodents such as streptozotocin–induced diabetic model [12]. To understand the pathogenesis of diabetic nephropathy and to develop preventive and therapeutic methods against it, suitable animal models for this disease have been needed.
A Novel Model of Severe Insulin–Dependent Diabetes
Recently we generated transgenic (TG) mice over expressing the mutant form (Thr286Asp) of CaMKIIα in pancreatic β–cells [13]. Western blot and immunehistochemical analyses showed that CaMKIIα protein was over expressed in pancreatic β–cells of the TG mice. Cell proliferation in the TG islets was severely impaired as assessed by in vivo BrdU labeling analysis. NF–?B accumulated in nuclei of the TGβ–cells at postnatal day (P) 21, which was associated with DNA laddering. The pancreatic insulin content was already significantly (p<0.01) lower in the TG mice than in the wild type (WT) mice at P7. The pancreatic insulin positive areas became negligible in the TG mice at P28 (Figures 2a–2c)). Serum insulin levels in the TG mice also decreased progressively along with aging, and were scarcely detectable at P28 (Figure 2d).

Figure 2: Immunohistochemical detection of insulin–producing pancreatic β cells in the representative sections of pancreas from WT mice (a) and the TG mice over expressing CaMKIIα (Thr286Asp) under the control of Insulin II promoter (b) at P28 (adopted from [13]). Percentages of insulin–positive area in whole pancreas section were determined at the indicated ages (c). Levels of insulin in serum in WT mice and the TG mice were determined at the indicated ages (d). The values are the means ± SE of at least four mice per group. ∗p< 0.05; ∗∗p< 0.01; ∗∗∗p< 0.001 compared with age-matched WT mice by Student’s t–test (adopted from [13]).
One hundred percent of TG mice developed severe hypoinsulinemic diabetes by P28. Blood glucose levels were significantly (p< 0.001) higher in the TG mice (44.4± 4.4mmol⁄l, n = 13) than in WT mice (8.9 ± 0.7mmol⁄l, n = 13) (Figure 3a).

Figure 3: Phenotypic characterization of WT mice and the CaMKIIa (Thr286Asp) TG mice at P140 (adopted from [13, 17]). (a) Blood glucose levels. (b) Urine albumin/creatinine ratio. (c) Mesangial area⁄ glomerular area. White bars and black bars indicate means± SE for WT mice and the TG mice, respectively. Statistical analyses were performed by Student’s t–test. ∗∗p<0.01; ∗∗∗p<0.001 compared with the values for age–matched WT mice. Macroscopic finding of kidneys from WT and the TG mice at P140. (d) PAS–staining of WT kidney. (e) and the TG kidney (f). Scale bar = 50 µm. Representative ERGs for maximal responses in WT mice. (g) and the TG mice. (h) at P140. The ERG signals (µV) were recorded at 0–150 ms after the flash.
Hemoglobin A1C levels were also significantly (p< 0.001) higher in the TG mice (9.4 ± 0.61%, n = 8) than in the WT mice (3.4 ± 0.08%, n = 13), suggesting that the TG mice will be valuable as a novel model of severe insulin–dependent DM.
Diabetic Nephropathy and Retinopathy in Cam Kinase Iiα TG Mice
The TG mice at P140–P168 developed severe renal and retinal lesions. Urinary albumin⁄ creatinine ratio (Figure 3b) as well as blood urea nitrogen (BUN) level were significantly elevated in the TG mice, indicating that the TG renal function is impaired. Histological analyses of kidneys revealed that body weight⁄kidney weight ratio, glomerular area, mesangial area, and mesangial⁄glomerular area ratio all increased in the TG mice (Figures 3c and 3d). The microscopic lesions noted by Periodic acid –Schiff (PAS) staining in the TG kidneys also consisted of glomerular hypertrophy, mesangial cell proliferation, mesangial expansion, and glomerulosclerosis (Figures 3e and 3f). Retinas of the TG mice demonstrated a dramatic loss of neuronal nuclei (NeuN)–positive nuclei, indicating a massive disruption of ganglion cells. Moreover, disorganization of the inner nuclear layer was evident in the TG retina. The corneal electro retinography (ERG) for maximal responses revealed waves each typical to a–waves, oscillatory potentials and b–waves for WT mice, but no discernible waves for the TG mice (Figures 3g and 3h).Thus, the TG mice were considered a good model of diabetic complications.
Diabetic Nephropathy and Inflammation
Macrophage infiltration has been reported to increase significantly in diabetic kidneys and thought to play significant roles in the progression of diabetic nephropathy [14,15]. There are at least two subtypes of resident macrophages in tissues [16]. One is referred to as M1 macrophages, which are classically activated by Th1 stimuli. M1 macrophages express high levels of proinflammatory cytokines and enhance tissue inflammatory response. Another is M2 macrophages, which are alternatively activated by Th2 stimuli. M2 macrophages express high levels of anti–inflammatory cytokines such as IL–10, and participate in the promotion of tissue repair, remodeling and vasculogenesis.
Diabetic Nephropathy and M1⁄M2 Macrophage
Expression of M1 macrophage markers such as CD11c, chemokine CCmotif receptor 2 (CCR2), tumor necrosis factor α (TNFα), interleukin–1β (IL–1β), monocyte chemotactic protein–1 (MCP–1), and nitric oxide synthase 2 (NOS2) in kidney was then analyzed. Among them, the mRNA levels of TNFα (1.77–fold) and MCP–1 (4.77–fold) were significantly higher (p < 0.05 for both) in the TG mice compared to WT mice (Figure 4a). M2 macrophage markers such as mannose receptor C–type1 (MRC1), chitinase 3–like 3 (CHI3l3), macrophage galactose N–acetyl–galactosamine specificlectin 2 (MGL2), CD209a, and interleukin–10 (IL–10) were next analyzed. Among them, the mRNA level of CHI3l3 (5.72–fold) wassignificantly higher (p < 0.001) in the TG mice compared to WT mice (Figure 4b). These results indicate that not only M1 macrophages but also M2 macrophages participate in the formation of diabetic nephropathy in the TG mice.
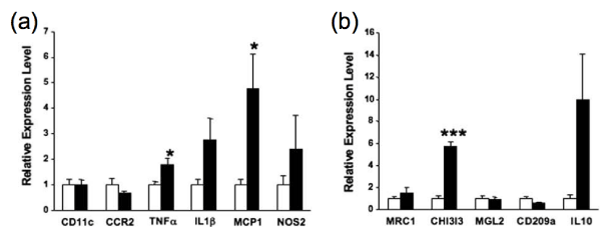
Figure 4: Quantitative real–time PCR analyses of macrophage marker mRNAs (a, b) (adopted from [17]). (a) mRNA levels of M1 macrophage markers (CD11c, CCR2, TNFα, IL1β, MCP1 and NOS2). (b) mRNA levels of M2 macrophage markers (MRC1, CHI3l3, MGL2, CD209a and IL10). In (a, b), the respective mRNA levels normalized to 18S ribosomal RNA (internal control) levels were shown. White bars and black bars indicate means ± SE for WT mice and TG mice, respectively. Statistical analyses were performed by Student’s t-test. ∗p<0.05; ∗∗∗p<0.001 compared with the values for age-matched WT mice.

Figure 5: Immunohistochemical detection of osteopontin (a, b) in the kidneys of WT mice (a) and TG mice (b) (adopted from [17]). Images are representative from studies of n=2 mice for each genotype. Scale bars = 100 µm.
Diabetic Nephropathy and Osteopontin
In order to identify genes whose expression levels were changed in the TG kidney, we carried out cDNA microarray analysis of mouse kidneys at P84. Genes with mean fold changes above 2.00 or less than0.60 were listed (Table).
![]()
Gene Name
Function
Gene Bank#
Fold
Up-regulated
cyclin D2
cell-cycle
NM_009829
6.70
sodium channel, nonvoltage-gated 1 beta
ion transport
NM_011325
4.40
nectin-lke 1
calcium ion binding
NM_053199
3.40
Notch-regulated ankyrin repeat protein
regulation of transcription
NM_025980
3.30
calcium channel, voltage-dependent, gamma subunit 3
calcium ion transport
NM_019430
3.21
pleckstrin homology domain containing, family B member 1
protein binding
NM_013746
3.18
RAS protein activator like 1
GTPase activator activity
NM_013832
3.12
receptor-interacting serine-threonine kinase 3
I?B kinase/ NF?B cascade
NM_019955
2.99
membrane-spanning 4-domains, subfamily A, member 6D
signal transduction
NM_026835
2.72
granzyme A
apoptosis
NM_010370
2.57
osteopontin
immune response
NM_009263
2.35
guanine nucleotide binding protein, alpha 11
G-protein signaling pathway
NM_010301
2.34
hydroxyacid oxidase 3
fatty acid metabolic process
NM_019545
2.23
lipopolysaccharide binding protein
lipid binding
NM_008489
2.15
interleukin 21 receptor
interleukin receptor activity
NM_021887
2.15
interleukin enhancer binding factor 3
protein amino acid methylation
NM_010561
2.06
catenin delta 2
cell adhesion
NM_008729
2.00
Down-regulated
tumor rejection antigen gp96
molecular chaperone
NM_011631
0.59
solute carrier family 21, member 1
renal organic anion transport
NM_013797
0.58
carbonic anhydrase 4
renal bicarbonate reabsorption
NM_007607
0.56
testis specific gene A2
sperm mobility
NM_025290
0.55
pleckstrin
T-cell activation
NM_019549
0.53
hydroxysteroid 11-beta dehydrogenase 1
cortisol metabolism
NM_008288
0.51
lipoprotein lipase
lipid transporter activity
NM_008509
0.473
lymphocyte antigen 6 complex, locus E
GPI anchor binding
NM_008529
0.460
Mpv17 transgene, kidney disease mutant-like
molecular function
NM_033564
0.421
Mouse ornithine decarboxylase
polyamine biosynthesis
NM_013614
0.348
DNA segment, Chr 7, Roswell Park 2 complex, expressed
hydrolase activity
NM_033080
0.332
hemoglobin, beta adult minor chain
iron ion binding
NM_016956
0.205
Table 1: Genes up– or down–regulated in CaMKIIα (Thr286Asp) TG mouse kidney (adopted from [17]).
The fold changes are mean comparisons between the TG kidney and WT kidney.
Osteopontin mRNA increased 2.35 fold in the TG kidney. Osteopontin is a chemokine–like, extracellular matrix–associated protein with diverse functions [18]. The elevated expression of osteopontin in renal cortex has also been reported in murine models of diabetes such as streptozotocin–induced diabetic rats [19] and db⁄db mice [20]. We performed immunohistochemical detection of osteopontin in the kidneys of TG and WT mice at P140. The glomeruli and the tubular epithelial cells were stained much more strongly forosteopontin in the TG kidney as compared to the WT kidney (Figures 5a and 5b). Osteopontin may recruit macrophages to renal cortex and induce inflammatory immune response.
Platelet Derived Growth Factor in Diabetic Nephropathy
Platelet–derived growth factor (PDGF) is a potent mitogen that stimulates extracellular matrix accumulation in mesangial cells [21]. PDGF family members, PDGF–A, PDGF–B, PDGF–C and PDGF–D, are assembled as disulphide–linked homo– or heterodimers [22]. These signals are mediated by two types of PDGF receptors (PDGFRs),PDGFR–α and PDGFR–β.
In the diabetic kidney, up regulation of the PDGF pathway has been shown in experimental diabetic nephropathy [23–25] and in the kidneys of patients with diabetes [26]. Among them, the production of PDGF–B and PDGFR–β is specifically increased and correlates to the progress of glomerular lesions such as diabetic nephropathy [27,28]. The in vitro exposure to high glucose also induces PDGF–B production in human proximal tubular epithelial cells and mesangial cells, and PDGFR–β production in mesangial cells [29–31]. A number of specific interventions aimed at neutralizing PDGF–Bor –D or blocking PDGFR–β have been shown to reduce mesangial cell proliferation and matrix accumulation and to ameliorate renaldysfunction in experimentally induced glomerulonephritis [32–35]. In contrast, the effects of anti–PDGF therapy on diabetic nephropathy have not been well characterized so far [36]. To our best knowledge, intervention studies involving PDGF are limited to the work showing that tyrosine kinase inhibition with 4–(4–Methylpiperazin– 1–ylmethyl)–N– [4–metthyl–3–(4–pyridin–3–ylpyrimidin–2–ylamino) phenyl] benzamide monomethanesulfonate (imatinib), a chemical tyrosine kinase inhibitor, retards the development of diabetic nephropathy in diabetic mice [37].
Camkiiα (Thr286Asp) Transgenic and Pdgfr–? Conditional Knockout Mice
Conditional Pdgfr–β knockout mice (actin–Cre–ERTM+⁄−–Pdgfr–β flox⁄flox mice), which express a fusion protein consisting of Cre recombinase and a mutated form of the mouse oestrogen receptor ligand–binding domain under the control of the actin promoter, were generated as described previously by Tokunaga et al [38]. By crossing the CaMKIIαTg⁄+ mice and the actin–Cre–ERTM+⁄−– Pdgfr–β flox⁄flox mice, CaMKIIαTg⁄+– actin–Cre–ERTM+⁄− Pdgfr–βflox⁄flox mice were generated. We examined the following four mouse strains with a CD–1 background: CaMKIIα+⁄+ –actin –Cre–ERTM−⁄− –Pdgfr–βflox⁄ flox mice (WT); CaMKIIα+⁄+ –actin –Cre–ERTM+⁄− – Pdgfr–βflox⁄flox mice (KO); CaMKIIαTg⁄+ –actin–Cre–ERTM−⁄− –Pdgfr–βflox⁄flox mice (DM); and CaMKIIαTg⁄+ –actin –Cre–ERTM+⁄− –Pdgfr–βflox⁄flox mice (DM–KO). All four strains of mice were similarly treated with tamoxifen at P28. The blood glucose levels of the DM and DM–KO mice increased markedly from P21, and reached a level higher than 44.4mmol⁄l thereafter. Deletion of Pdgfr–β did not affect body weight, blood glucose levels or blood pressure in either normoglycaemic control mice or diabetic CaMKIIα transgenic mice [39].

Figure 6: Deletion of Pdgfr–β improved diabetic nephropathy in DM mice [39]. (a) Urinary albumin⁄creatinine ratio at the indicated days of age. (b) Mesangial area⁄glomerular area ratio at P112. Data shown are the means±SE of at least six mice per group. ∗p<0.05; ∗∗p<0.01 (c) Urinary 8–OHdG levels at P112. Data shown are the means±SE of at least six mice per group. ∗p<0.05; ∗∗p<0.01 (d) Representative images of western blotting of renal cortex with anti–NOX4 and anti–β–action antibodies.∗p<0.05; ∗∗p<0.01.
Diabetic Nephropathy in Camkiiα (Thr286Asp) Transgenic and Pdgfr–? Conditional Knockout Mice
The urinary albumin to creatinine ratio was higher in the DM micethan in the WT mice at P112. The deletion of Pdgfr–βdecreased the ratio significantly in diabetic CaMKIIα transgenic mice (Figure 6a). Deletion of Pdgfr–β improved the pathological changes in glomeruli observed in CaMKIIα transgenic mice. Microscopic examinations demonstrated that the glomeruli of DM mice were significantly larger than those in WT mice. They frequently showed the sclerotic changes of glomeruli, such as segmental changes at P112. The mesangial area to glomerular area ratio in DM–KO mice was significantly lower than in DM mice (Figure 6b).
The urinary 8–hydroxydeoxyguanosine (8–OHdG) level measured as oxidative stress marker was significantly higher in DM mice than in WT mice, and was significantly lower in DM–KO mice than in DM mice at P112 (Figure 6c). The production of NAD(P)H oxidase 4 (NOX4), a renal homologue of NAD(P)H oxidase, tended to be up regulated in DM mice, and significantly down regulated in DM–KO mice (Figure 6d).
It is reported that NOX4 is activated by PDGF–B followed by the generation of reactive oxygen species [40,41]. Inhibition of NOX4 ameliorates diabetic nephropathy with decreasing NAD (P) H–dependent reactive oxygen species generation [42]. These reports suggest that down regulation of NOX4 may be involved, at least partly,in the improvement of diabetic nephropathy observed in DM–KO mice. Our present study suggests that enhanced PDGFR–β signaling plays important roles in the development of diabetic nephropathy in vivo and that increased oxidative stress is involved in this process
Conclusions
The CaMKIIα (Thr286Asp) transgenic mice established here in would be valuable as a novel model of severe insulin–dependent diabetes accompanied by an early progression of diabetic micro vascular complications.
Acknowledgements
This work was supported by a Grant–in–Aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture, Japan (18209033 and 21591126 to K. Tobe, 20390108 to M. Sasahara and 22590971 to I. Usui), a Grant–in–Aid for the 21st Century COE Program from the Ministry of Education, Culture, Sports, Science and Technology, Japan and the Research Fund from the Japan Diabetes Foundation. We thank S. Fujisaka, S. Senda and A. Mahmood for excellent technical assistance and useful discussion.
References
- Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004; 279: 42351-42354.
- Brownlee M. A radical explanation for glucose-induced Β cell dysfunction. J Clin Invest. 2003; 112: 1788-1790.
- Krolewski M, Eggers PW, Warram JH. Magnitude of end-stage renal disease in IDDM: a 35 year follow-up study. Kidney Int. 1996; 50: 2041-2046.
- Steinke JM, Mauer M. Lessons learned from studies of the natural history of diabetic nephropathy in young type 1 diabetic patients. Pediatr Endocrinol Rev. 2008; 5 Suppl 4: 958-963.
- Takasawa S, Tohgo A, Noguchi N, Koguma T, Nata K, et al. Synthesis and hydrolysis of cyclic ADP-ribose by human leukocyte antigen CD38 and inhibition of the hydrolysis by ATP. J Biol Chem. 1993; 268: 26052-26054.
- Takasawa S, Nata K, Yonekura H, Okamoto H. Cyclic ADP-ribose in insulin secretion from pancreatic beta cells. Science. 1993; 259: 370-373.
- Lee HC, Aarhus R, Graeff R, Gurnack ME, Walseth TF. Cyclic ADP ribose activation of the ryanodine receptor is mediated by calmodulin. Nature. 1994; 370: 307-309.
- Takasawa S, Ishida A, Nata K, Nakagawa K, Noguchi N, et al. Requirement of calmodulin-dependent protein kinase II in cyclic ADP-ribose-mediated intracellular Ca2+ mobilization. J Biol Chem. 1995; 270: 30257-30259.
- Takasawa S, Akiyama T, Nata K, Kuroki M, Tohgo A, et al. Cyclic ADP-ribose and inositol 1,4,5-trisphosphate as alternate second messengers for intracellular Ca2+ mobilization in normal and diabetic Β-cells. J Biol Chem. 1998; 273: 2497-2500.
- Takasawa S, Kuroki M, Nata K, Noguchi N, Ikeda T, et al. A novel ryanodine receptor expressed in pancreatic islets by alternative splicing from type 2 ryanodine receptor gene. Biochem Biophys Res Commun. 2010; 397: 140-145.
- Doi T, Hattori M, Agodoa LY, Sato T, Yoshida H, et al. Glomerular lesions in nonobese diabetic mouse: before and after the onset of hyperglycemia. Lab Invest. 1990; 63: 204-212.
- Williamson JR, Chang K, Tilton RG, Prater C, Jeffrey JR, et al. Increased vascular permeability in spontaneously diabetic BB/W rats and in rats with mild versus severe streptozocin-induced diabetes. Prevention by aldose reductase inhibitors and castration. Diabetes. 1987; 36: 813-821.
- 13. Kato I, Oya T, Suzuki H, Takasawa K, Ichsan AM, et al. A novel model of insulin-dependent diabetes with renal and retinal lesions by transgenic expression of CaMKIIa (Thr286Asp) in pancreatic ß-cells. Diabetes Metab Res Rev. 2008; 24: 486-497.
- Chow F, Ozols E, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int. 2004; 65: 116-128.
- Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrol Dial Transplant. 2004; 19: 2987-2996.
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003; 3: 23-35.
- Suzuki H, Kato I, Usui I, Takasaki I, Tabuchi Y, et al. Characterization of diabetic nephropathy in CaM kinase IIa (Thr286Asp) transgenic mice. Biochem Biophys Res Commun. 2009; 379: 38-42.
- Denhardt DT, Guo X. Osteopontin: a protein with diverse functions. FASEB J. 1993; 7: 1475-1482.
- Fischer JW., Tschope C, Reinecke A, Giachelli CM, Unger T. Upregulation of osteopontin expression in renal cortex of streptozotocin-induced diabetic rats is mediated by bradykinin. Diabetes. 1998; 47: 1512-1518.
- Susztak K, Bottinger E, Novetsky A, Liang D, Zhu Y, et al. Molecular profiling of diabetic mouse kidney reveals novel genes linked to glomerular disease. Diabetes. 2004; 53: 784-794.
- Johnson RJ, Raines EW, Floege J, Yoshimura A, Pritzl P, et al. Inhibition of mesangial cell proliferation and matrix expansion in glomerulonephritis in the rat by antibody to platelet-derived growth factor. J Exp Med. 1992; 175: 1413-1416.
- Tallquist M, Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004; 15: 205-213.
- #23. Kelly DJ, Gilbert RE, Cox AJ, Soulis T, Jerums G, et al. Aminoguanidine ameliorates overexpression of prosclerotic growth factors and collagen deposition in experimental diabetic nephropathy. J Am Soc Nephrol. 2001; 12: 2098-2107.
- Nakagawa H, Sasahara M, Haneda M, Koya D, Hazama F, et al. Immunohistochemical characterization of glomerular PDGF B-chain and PDGF beta-receptor expression in diabetic rats. Diabetes Res Clin Pract. 2000; 48: 87-98
- Nakamura T, Fukui M, Ebihara I, Osada S, Nagaoka I, et al. mRNA expression of growth factors in glomeruli from diabetic rats. Diabetes. 1993; 42: 450-456.
- Langham RG, Kelly DJ, Maguire J, Dowling JP, Gilbert RE, et al. Over-expression of platelet-derived growth factor in human diabetic nephropathy. Nephrol Dial Transplant. 2003; 18: 1392-1396.
- Matsuda M, Shikata K, Makino H, Sugimoto H, Ota K, et al. Gene expression of PDGF and PDGF receptor in various forms of glomerulonephritis. Am J Nephrol. 1997; 17: 25-31.
- Uehara G, Suzuki D, Toyoda M, Umezono T, Sakai H. Glomerular expression of platelet-derived growth factor (PDGF)-A, -B chain and PDGF receptor-a, -ß in human diabetic nephropathy. Clin Exp Nephrol. 2004; 8: 36-42.
- Di Paolo S, Gesualdo L, Ranieri E, Grandaliano G, Schena FP. High glucose concentration induces the overexpression of transforming growth factor-ß through the activation of a platelet-derived growth factor loop in human mesangial cells. Am J Pathol. 1996; 149: 2095-2106.
- Doi T, Vlassara H, Kirstein M, Yamada Y, Striker GE, et al. Receptor-specific increase in extracellular matrix production in mouse mesangial cells by advanced glycosylation end products is mediated via platelet-derived growth factor. Proc Natl Acad Sci U S A. 1992; 89: 2873-2877.
- Inaba T, Ishibashi S, Gotoda T, Kawamura M, Morino N, et al. Enhanced expression of platelet-derived growth factor-beta receptor by high glucose. Involvement of platelet-derived growth factor in diabetic angiopathy. Diabetes. 1996; 45: 507-512.
- Ostendorf T, Kunter U, van Roeyen C, Dooley S, Janjic N, et al. The effects of platelet-derived growth factor antagonism in experimental glomerulonephritis are independent of the transforming growth factor-ß system. J Am Soc Nephrol. 2002; 13: 658-667.
- Ostendorf T, Rong S, Boor P, Wiedemann S, Kunter U, et al. Antagonism of PDGF-D by human antibody CR002 prevents renal scarring in experimental glomerulonephritis. J Am Soc Nephrol. 2006; 17: 1054-1062.
- Takahashi T, Abe H, Arai H, Matsubara T, Nagai K, et al. Activation of STAT3/Smad1 is a key signaling pathway for progression to glomerulosclerosis in experimental glomerulonephritis. J Biol Chem. 2005; 280: 7100-7106.
- Yagi M, Kato S, Kobayashi Y, Kobayashi N, Iinuma N, et al. Beneficial effects of a novel inhibitor of platelet-derived growth factor receptor autophosphorylation in the rat with mesangial proliferative glomerulonephritis. Gen Pharmacol. 1998; 31: 765-773.
- Floege J, Eitner F, Alpers CE. A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol. 2008; 19: 12-23.
- 37. Lassila M, Jandeleit-Dahm K, Seah KK, Smith CM, Calkin AC, et al. Imatinib attenuates diabetic nephropathy in apolipoprotein E-knockout mice. J Am Soc Nephrol. 2005; 16: 363-373.
- Tokunaga A, Oya T, Ishii Y, Motomura H, Nakamura C, et al. PDGF receptor beta is a potent regulator of mesenchymal stromal cell function. J Bone Miner Res. 2008; 23: 1519-1528.
- Suzuki H, Usui I, Kato I, Oya T, Kanatani Y, et al. Deletion of platelet-derived growth factor receptor-ß improves diabetic nephropathy in Ca2+/calmodulin-dependent protein kinase IIa (Thr286Asp) transgenic mice. Diabetologia. 2011; 54: 2953-2962.
- De Minicis S, Bataller R, Brenner DA. NADPH oxidase in the liver: defensive, offensive, or fibrogenic? Gastroenterology. 2006; 131: 272-275.
- Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008; 57: 1446-1454.
- Fujii M, Inoguchi T, Maeda Y, Sasaki S, Sawada F, et al. Pitavastatin ameliorates albuminuria and renal mesangial expansion by downregulating NOX4 in db/db mice. Kidney Int. 2007; 72: 473-480.