
Case Report
Austin J Endocrinol Diabetes. 2024; 11(1): 1104.
Tailored Care for Uncommon Neonatal Hyponatremia: Collaborative Approach by Multidisciplinary Experts
Bogdan Mihai Pascu1,2*; Vasilica Plaiasu3; Lucia Rosu2,4; Cristina Stoica5; Elena-Emanuela Stan; Alexandra Jafal
1Department of Endocrinology, National Institute for Mother and Child Health “Alessandrescu-Rusescu”, Romania
2Department of Faculty of Medicine, Carol Davila University of Medicine and Pharmacy, Romania
3Department of Genetic, National Institute for Mother and Child Health “Alessandrescu-Rusescu”, Romania
4Department of Pediatric, National Institute for Mother and Child Health “Alessandrescu-Rusescu”, Romania
5Department of Nephrology, Fundeni Clinical Institute, Romania
*Corresponding author: Bogdan Mihai Pascu Department of Endocrinology, National Institute for Mother and Child Health “Alessandrescu-Rusescu”, Romania. Email: drpascu@endocrinologiepediatrica.ro
Received: March 22, 2024 Accepted: April 23, 2024 Published: April 30, 2024
Abstract
Aldosterone Synthase Deficiency (ASD) is a rare cause of hyponatremia, commonly found in infancy and if is early diagnosed it has a very good prognosis. ASD has been classified in 2 types, type 1, which is the rarest form and type 2. Patients with ASD usually present with salt-wasting, insufficient release of potassium in the urine, metabolic acidosis and failure to thrive in early infancy. This paper reports on one ASD case of a 6 weeks old female who presented with salt-wasting crisis, hyperkalemia, metabolic acidosis and failure to thrive. During the initial Intensive Care Unit (ICU) admission phase, ASD was not suspected due to nonspecific signs. During the second admission in the ICU, two weeks after the initial admission, guided by the pediatric endocrinologist, empiric fludrocortisone therapy was started with improvements in weight gain. Genetic testing identified two heterozygous pathogenic and likely pathogenic variants in the CYP11B2 gene, which confirmed corticosterone methyloxidase deficiency. Plasma steroid profile showed high plasma renine activity, inappropriate normal aldosterone secretion and normal 18-hydroxycorticosterone, compatible with type 1 aldosterone synthase deficiency. Through this study we aim to underline the importance of early recognizing this life-threatening pathology, by hormonal investigations, followed by genetic testing to facilitate the clinical management of the patient.
Keywords: Aldosterone synthase deficiency; Salt-wasting; Failure to thrive; Hyperreninemia
Introduction
Aldosterone synthase is expressed in the adrenal cortex and catalyses the final three steps of the aldosterone biosynthesis: from 11-deoxycorticosterone to corticosterone, then to 18-hydroxycorticosterone and finally to aldosterone [1]. Aldosterone synthase deficiency is a rare autosomal recessive disorder [2], that has an unknown prevalence [1], characterized by excessive sodium release in the urine, associated with insufficient release of potassium in the urine, usually starting in infancy [3]. This condition has a potential life-threatening risk, so it is necessary to conduct hormonal investigations, along with genetic testing in suspected patients to facilitate clinical management [2]. Aldosterone synthase deficiency is classified in type 1 and type 2 depending on the production of 18-hydroxycorticosterone, which is increased only in type 2 ASD. Aldosterone synthase deficiency type 1 occurs less frequently and typically results in low or absent aldosterone production and is more severe than ASD type 2 [4]. Aldosterone synthase deficiency manifests in the first weeks of life with nausea, vomiting, feeding problems and failure to thrive. Clinical severity improves with age, patients being frequently asymptomatic during adulthood despite not receiving mineralocorticoid therapy [1].
Case Presentation
We describe a 6-week-old infant, female, who came from a pregnancy obtained via IVF, with physiological evolution, delivered via cesarean section, at 38 weeks gestational age, with a birth weight of 3070 g, birth length of 51 cm and an Apgar Score of 10, who was breastfed combined with formula one month, then switched to formula only. After birth, she had an extended neonatal metabolic screening (including cystic fibrosis), which was negative. Both her parents were healthy, with no chronic illnesses. From the patient’s medical history, we found that she presented first at the pediatrician at 2 weeks old, for not regaining her birth weight (2900 g at 2 weeks) and breast enlargement, so she was referred to the endocrinologist. Four weeks later, she came back to the pediatric department, where the pediatrician concluded that she gained only 280 g in 6 weeks and he also collected an arterial blood gas test, which showed hyponatremia (125 mmol/l) and hyperkalemia (6.5 mmol/l), so she was referred to a multidisciplinary care unit for further investigations. At the first evaluation in our clinic, clinical examination showed mottled skin, systolic murmur grade II/VI, mildly bloated abdomen and axial hypotonia and laboratory tests showed hyponatremia (127 mmol/l), hyperkalemia (6.5 mmol/l) and the 24 h urine collection showed that sodium was 60 mmol/l, potassium was 14.50 mmol/l and urine density was 1005. Additional investigations were made and at the cardiac ultrasound we found out that she had patent foramen ovale, with a left-right shunt and small persistent ductus arteriosus. During neurologic evaluation, the pediatric neurologist concluded that the infant had axial hypotonia and mild left-sided plagiocephaly. During hospitalization, the infant received age-appropriate formula feeding, in progressively increasing amounts, hydroelectrolytic rebalancing infusion and oral sodium chloride 5.85%, with favorable evolution and good weight gain (330g/10 days).
Two weeks later, she came back to the emergency room with decreased appetite and one episode of vomiting. Laboratory investigations were repeated and the electrolyte test showed hyponatremia (128 mmol/l), hyperkalemia (6.50 mmol/l) and hypercalcemia (1.48 mmol/l), 17-hydroxyprogesterone was in the normal range (1.27 ng/ml), so we excluded the majority of congenital adrenal hyperplasia forms and the thyroid function showed that the infant had subclinical hypothyroidism. During hospitalization, sequential blood gas analyses were collected revealing persistent hyponatremia and hyperkalemia (Table 1).
The pediatric endocrinologist recommended additional tests for the hypothalamic-pituitary-adrenal (HPA) axis - cortisol and ACTH were in the normal range, excluding primary adrenal insufficiency, aldosterone level had inappropriately normal value (14.9 ng/dl), and renin value was above the detection limit (> 5000 uUI/ml), the fasting blood glucose was in the normal range (84 mg/dl), plasmatic osmolality was 259.11 mOsm/kg and the urine osmolality was inappropriately high (207.38 mOsm/kg). The endocrinologist requested additional pediatric nephrology evaluation and genetic counselling (searching for causes of aldosterone synthase (CYP11B2) deficiency/aldosterone resistance) and started empiric fludrocortisone therapy.
During nephrology evaluation, a renal ultrasound was performed and there were no features of nephrocalcinosis. Based on laboratory findings, a tubulopathy of Gitelman syndrome or Bartter syndrome was ruled out, which could have been suggestive in the context of growth failure and hyperreninemia. Both pediatric endocrinologist and nephrologist strongly suggested genetic testing (WES).
The geneticist concluded that the patient had a weight gain, but she was only on the 5th percentile, she was alert, cried vigorously and had axial hypotonia. The geneticist also recommended genetic testing (WGS/WES). Genetic testing was performed, and two heterozygous pathogenic and likely pathogenic variants were identified in the CYP11B2 gene. If variants in the compound heterozygous state (in trans), the genetic diagnosis of autosomal recessive corticosterone methyloxidase deficiency is confirmed.
The infant was reevaluated at the endocrinology department and the laboratory tests found that cortisol and electrolyte test had normal values and the renine value dropped at 648.2 uUI/ml, so it was possible to calculate the aldosterone-to-renine ratio, which was low (0.009), suggestive for primary hypoaldosteronism. The endocrinologist also requested the 18-hydroxycorticosterone value, which was normal, suggestive for ASD type 1.
During the management process of this case of hyponatremia, we tried to exclude other endocrine causes like congenital adrenal hyperplasia, primary adrenal insufficiency, cerebral salt wasting syndrome or inappropriate anti-diuretic hormone secretion, renal causes like tubulopathy of Gitleman or Barter syndromes, nephrotic syndrome and gastrointestinal causes (Figure 1) [5].
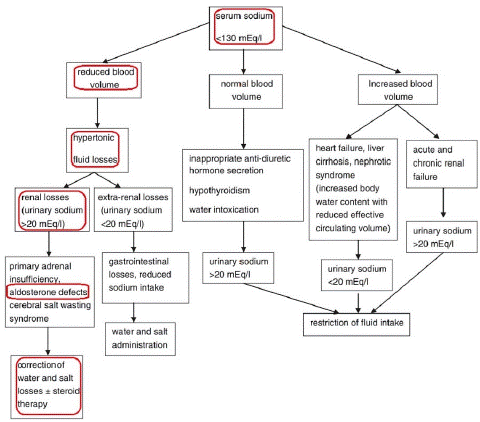
Figure 1: Differential diagnosis and treatment of hyponatremia in infancy [5].
In our patient, treatment with oral sodium supplementation and fludrocortisone was initiated in October 2023, with progressive increasing doses and the infant rapidly started to catch up growth (Figure 2).
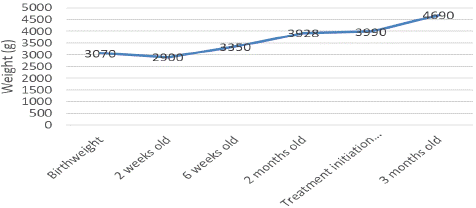
Figure 2: Weight evolution from birth to 3 months old.
At 6 months old, the endocrinology reevaluation showed the evolution of the renine value which became normal after approximately 3 months of treatment (Figure 3&4) .
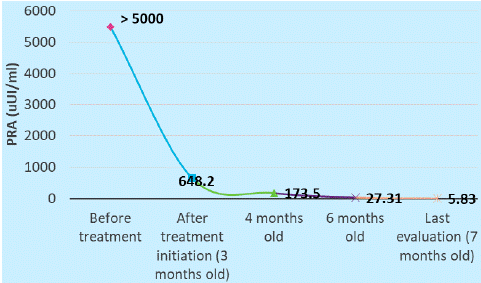
Figure 3: Renine evolution.
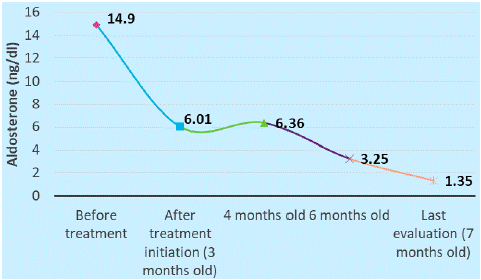
Figure 4: Aldosterone evolution.
Discussions
We described a 6-week-old infant who presented with a salt-wasting crisis, failure to thrive and feeding difficulties. The clinical presentation along with typical biochemical alterations like hyponatremia, hyperkalemia, high plasma renin levels and inappropriate normal aldosterone suggested the underlying diagnosis, which was confirmed by genetic analysis.
In infancy, clinical presentation can be dramatic, with significant hemodynamic impact and life-threatening episodes of seizures and coma. Usually, this condition improves with age, adults being frequently asymptomatic despite no mineralocorticoid therapy, this fact being associated with a gradual declining dependency on aldosterone during lifetime for the normal ionic imbalance due to possible extra-adrenal compensatory mechanisms and alternative ACTH-dependent pathways for mineralocorticoid biosynthesis [1].
It is important to underline that high aldosterone plasma levels in infancy do not rule out aldosterone insufficiency and might mislead differential diagnosis with pseudohypoaldosteronism. Therapeutic requests and growth impairment in hypoaldosteronism vary even with a common genetic background [6].
The aldosterone synthase enzyme is involved in a series of three chemical reactions that produce aldosterone from other precursor molecules [3] (Figure 5).
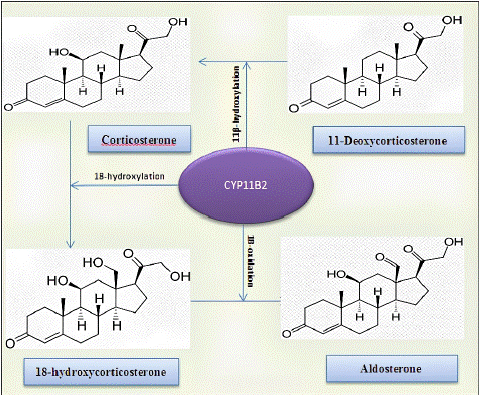
Figure 5: Enzymatic reactions catalyzed by CYP11B2 enzyme [7].
ASD has been classified in [2]:
Type 1 – the enzymatic CYP11B2 activity is completely abolished, with low to normal levels of 18-OHB [2];
Type 2 – the mutation in the CYP11B2 gene only decreases 18-hydroxylase and 18-oxidase activities, but not 11 Β-hydroxylase activity and 18-OHB levels are markedly elevated [2].
In our case, after dosing 18-OHB, which had a normal value, type 1 ASD deficiency, which is rarer, was confirmed.
Conclusions
Aldosterone synthase deficiency is a rare autosomal recessive disorder, characterized by a disruption in aldosterone synthesis, that clinically manifests in salt-wasting crisis, failure to thrive and feeding difficulties, in infancy. It is important to recognize this condition as it has a good long-term prognosis when adequate fludrocortisone and oral sodium supplementation is instituted. Therefore, genetic confirmation in suspected adults and their partners is essential in assessing the genetic implications in their offspring and to minimize the potentially life-threatening risks of this condition.
In our case, fludrocortisone therapy administered in progressively increasing doses associated with oral sodium supplementation determined weight gain and resolution of the biochemical abnormalities.
Author Statements
Conflict of Interest
The authors declare no conflict of interest.
References
- Lages AS, Vale B, Oliveira P, Cardoso R, Dinis I, Carrilho F, et al. Congenital hyperreninemic hypoaldosteronism due to aldosterone synthase deficiency type I in a Portuguese patient - Case report and review of literature. Arch Endocrinol Metab. 2019; 63: 84-88.
- Hui E, Yeung MC, Cheung PT, Kwan E, Low L, Tan KC, et al. The clinical significance of aldosterone synthase deficiency: report of a novel mutation in the CYP11B2 gene. BMC Endocr Disord. 2014; 14: 29.
- MedlinePlus. Bethesda (MD): National Library of Medicine (US); Corticosterone methyloxidase deficiency. 2024.
- Karen Lin-Su, Oksana Lekarev, Maria I. New, Chapter 87 - Genetic Disorders of the Adrenal Gland, Editor(s): David Rimoin, Reed Pyeritz, Bruce Korf, Emery and Rimoin’s Principles and Practice of Medical Genetics (Sixth Edition), Academic Press. 2013; 1-37.
- Bizzarri Carla, Olivini Nicole, Pedicelli Stefania, Marini Romana, Giannone Germana, Cambiaso Paola, et al. Congenital primary adrenal insufficiency and selective aldosterone defects presenting as salt-wasting in infancy: a single center 10-year experience. Italian Journal of Pediatrics. 2024; 42: 73.
- Martín-Rivada Á, Argente J, Martos-Moreno GÁ. Aldosterone deficiency with a hormone profile mimicking pseudohypoaldosteronism. J Pediatr Endocrinol Metab. 2020 Nov 26;33(11):1501-1505.
- Tarek Abdel Ghafar M. Aldosterone Synthase Gene (CYP11B2) Polymorphisms and Enhanced Cardiovascular Risk. The Recent Topics in Genetic Polymorphisms. Intech Open. 2020. 2024.