
Case Series
Austin J Endocrinol Diabetes. 2024; 11(1): 1107.
Resistant Rickets: A Case Series of Six Different Etiologies
Sayantan Chakraborty¹*; Debaditya Das²; Piyas Gargari²; Anish Kar²; Sujoy Ghosh³; Pradip Mukhopadhyay³
1Post-Doctoral Trainee, Department of Endocrinology, IPGME & R, Kolkata, India
2Senior Resident, Department of Endocrinology, IPGME & R, Kolkata, India
3Professor, Department of Endocrinology, IPGME & R, Kolkata, India
*Corresponding author: Sayantan Chakraborty, Post-Doctoral Trainee, Department of Endocrinology, IPGME & R, Kolkata, India. Email: sc9163@gmail.com
Received: July 22, 2024 Accepted: August 09, 2024 Published: August 16, 2024
Abstract
Rickets is a metabolic bone disorder characterized by defective mineralization of epiphyseal growth plate due to various disorders of calcium and phosphate metabolism. Resistant rickets is defined by persistence of clinical, biochemical or radiological evidence of rickets even after treating with adequate dose of vitamin D3 for 8-12 weeks. It has various causes which is broadly classified into calcipenic variant and phosphopenic variant. Calcipenic rickets should be suspected in those patients having defect in vitamin D metabolism. In this case series, case no 1 and case no 4 presents two important cause of calcipenic vitamin d resistant rickets. Other variant of resistant rickets is phosphopenic rickets which is broadly divided into FGF23 dependent and independent causes. Case no 2 and case no 6 presents two different causes of FGF23 dependent diseases where as case no 3 and case no 5 presents two manifestations of FGF23 independent phosphopenic rickets. Thorough investigations should be performed to diagnose these cases and appropriate treatment and monitoring is necessary to ameliorate these group of patients.
Keywords: Rickets; Calcipenic rickets; Phosphopenic rickets; Renal tubular acidosi
Introduction
Rickets is a metabolic bone disorder characterized by defective mineralization of epiphyseal growth plate due to various disorders of calcium and phosphate metabolism. Worldwide most common cause of rickets is Vitamin D deficiency, which can be managed with high dose vitamin D supplementation for 8 to 12 weeks. But few patients remain refractory to treatment with high dose of vitamin D collectively termed as Vitamin D resistant rickets. It has various etiologies. In this article we will present a series of six cases of resistant rickets caused due to different etiologies.
Case 1
4 years old female child presented with bowing of legs and swelling of ankle (Figure 1) and wrist since 18 months of age, pain over both legs during walking since last 2 years and total loss of scalp hair for last 6 months. No history of tetany, convulsion, delayed dentition, fracture, dental abscess. On evaluation she was found to have hypocalcemia, normal 25(OH) vit D and serum phosphate, elevated PTH and very high alkaline phosphatase and 1,25 dihydroxyvitamin D. There was presence of cupping, fraying over knee joint on radiograph. Based on clinical, biochemical and radiological findings provisional diagnosis of Vitamin D dependent ricket type 2 (VDDR 2) was made. Child was put on high dose elemental calcium (50mg/kg) and high dose calcitriol (40ng/kg) Currently, she is better with resolution of pain and she is under regular follow up.
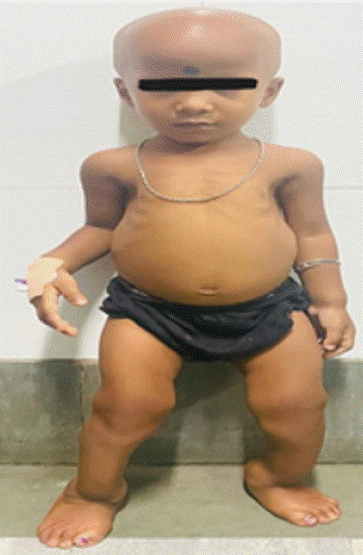
Figure 1: Alopecia and bowing of legs.
Case 2
3 years old male child presented with difficulty in walking since 1 year of age and progressive bowing of both lower limbs since 1.5 years of age which was associated with pain in both leg during walking (Figure 2). There was history of delayed dentition, dental caries but no history of fracture, alopecia, proximal muscle weakness. On examination he was found to have dolicocephaly, frontal bossing, dental carries, Harrison sulcus, widening of wrist, bilateral genu varus. On evaluation he was found to have prominent hypophosphatemia, normocalcemia, significant renal phosphate wasting (low TMP GFR) and elevated FGF 23 (Table 1) Based on clinical and biochemical findings whole exome sequencing was planned which revealed inactivating PHEX gene mutation (162 bp deletion in exon 3). Diagnosis of X linked hypophosphatemic rickets was established. Currently child is on oral phosphate sachet (40mg/kg/day) and calcitriol supplementation (30ng/kg/day). His biochemical parameters improved (normalization of alkaline phosphatase and improvement of serum phosphate level).
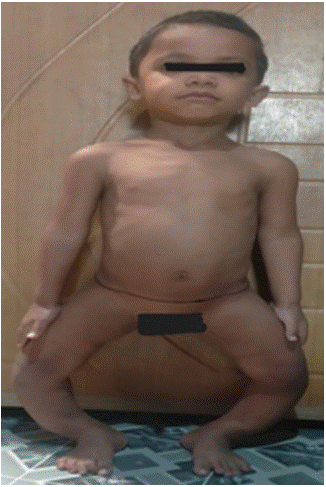
Figure 2: Progressive bowing of both lower limbs.
Case 3
3 years old girl child presented with failure to thrive, increased fatigability, lower limb deformity. Clinical examination revealed pallor, short stature, wrist widening and genu valgum without any hepatosplenomegaly Baseline biochemical investigation showed anaemia, hypophosphatemia, hypokalemia and metabolic acidosis (Table 1). Serum 25OH vitamin D was within normal limits, parathyroid hormone was normal. Urine analysis showed proteinuria, glycosuria, renal phosphate wasting as well as increased urinary β2 macroglobulin excretion indicating low molecular weight proteinuria. Skiagram of both wrists revealed cupping, fraying and splaying of metaphysis (Figure 3). She was diagnosed with generalized Proximal renal tubular dysfunction (fanconi syndrome). Evaluation was started for secondary causes of fanconi syndrome. Wilson disease, Galactosemia, Cystinosis had been ruled out after appropriate testing. Subsequent blood tandem mass spectrometry showed increased level of succinyl choline and urine gas chromatography and mass spectrometry showed increased excretion of 4 hydroxyphenyl acetic acid and hydroxyphenyl acetic acid, suggestive of type 1 Tyrosinemia. She was put on alkali therapy (4meq/kg/day) with phosphate (30mg/kg) supplementation. Currently she is clinically better with biochemical improvement of acidosis and hypokalemia.
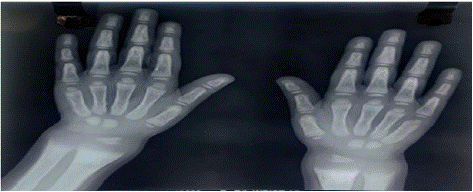
Figure 3: Cupping and fraying over wrist.
Case 4
2 years old boy presented with gradually progressive skeletal deformity in all 4 limbs for last 9 months. There was a history of hospital admission at 3 months of age with features of failure to thrive and recurrent vomiting, which was eventually diagnosed as DKA, treated with IV fluids and intravenous insulin. Post discharge, subcutaneous insulin was continued till 6 months of age after which the parents discontinued insulin therapy as he was having episodes of hypoglycemia. Post discontinuation his CBG levels remained within range. Clinical examination revealed dysmorphic facial features like hypertelorism, retrognathia, open anterior fontanelle with bilateral genu varum. On evaluation he was found to have hypocalcemia, hyperphosphatemia, raised alkaline phosphatase, low 1,25 dihydroxy vitamin D3 and elevated parathyroid hormone (Table 1). X ray revealed metaphyseal and epiphyseal rachitic changes (Figure 4). Contemplating a syndromic association, clinical exome sequencing was performed which showed pathogenic homozygous mutation in CYP27B1 gene on chromosome 12 and heterozygous mutation in ABCC8 gene on chromosome 11 thus explaining both skeletal changes and transient diabetes documented at infancy. He was symptomatically improved with Calcitriol (20ng/kg/day) therapy and no episodes of hyperglycemic emergency was documented till now.
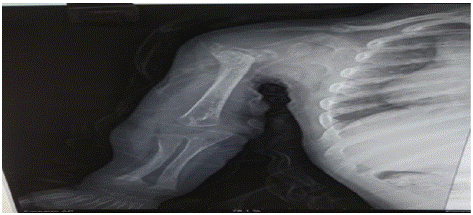
Figure 4: Epiphyseal and metaphyseal changes.
Case no 5
7 months old male child presented with bending of legs and swelling of ankle and wrists as noticed by mother. There was no history of any trivial trauma fractures, tetany, convulsion. On evaluation he was found to have hypophosphatemia, normal 25OH vitamin D and very high alkaline phosphatase, 1,25 OH vitamin D (Table 1). There was evidence of renal phosphate wasting (low TMP GFR) and suppressed FGF23 level, suppressed parathyroid hormone. Ultrasonography revealed no abnormality in both kidneys. On the basis of elevated 1 25 OH vitamin D and low TMP GFR, non FGF 23 mediated cause of renal phosphate wasting was established. Finally Whole exome sequencing report revealed autosomal recessive mutation in SLC34A3 leading to defective renal sodium dependent phosphate reabsorption through NPT2c in proximal tubule. He was treated with oral phosphate supplementation (40mg/kg) and currently is maintaining low normal serum phosphate.
![]()
Parameters
Case 1
Case 2
Case 3
Case 4
Case 5
Case 6
Calcium (8.8-10.2 mg/dl)
6.6
9.0
8.9
6.55
8.99
8.7
Phosphate (4.5-5.5 mg/dl)
3.4
2.1
1.5
4.64
2.0
2.4
TMP GFR (3.25-5.51 mmol/l)
2.25
1.3
1.45
1.6
ALP (upto 150 U/L)
2030
585
1796
497
843
25 OH vit D (30-100 ng/ml)
75
25
45
74
33
1,25 OH vit D (47-197 ng/ml)
>200
34
33
154
10
Intact PTH (35-55pg/ml)
1185
104
29
218
<3
26
FGF23 (18-108 pg/ml)
112
5.28
54
Blood pH
7.41
7.42
7.30
7.38
7.40
7.36
Bicarbonate (18-24mmol/l)
21
26
16
22
24
21
Table 1: Biochemical parameters of all patients.
Case no 6
13 years old female presented with pain in both legs and thigh and progressive bowing of both legs for last 5 years. There was history of toothache since childhood and spontaneous extraction of 5 teeth in last 3 years. On examination she was found to have widening of wrist, intorsion of both tibia and forefoot, bilateral genu varum. On evaluation she was found to have severe hypophosphatemia, significant phosphaturia (low TMP GFR) and non-suppressed FGF 23 level suggesting FGF 23 mediated hypophosphatemic rickets (Table 1). Genetic test could not be possible because of financial problems. She was managed with Calcitriol (30ng/kg) and elemental phosphate (40mg/kg) supplementation like other hypophosphatemic rickets. Her pain subsided with therapy. Currently she is maintaining low normal serum phosphate and under periodic follow up.
Discussion
Vitamin D resistant ricket is a less commonly encountered entity as compared with vitamin D deficiency rickets. But any child presenting with suspicion of resistant rickets should be thoroughly investigated. They are broadly divided into predominantly calcipenic and phosphopenic variant. Although profound hypocalcemia and hypocalcemic symptoms are common in calcipenic variant and profound hypophosphatemia and dental abnormality are common in phosphopenic variant, best parameter to differentiate between the two is serum parathyroid hormone level which will be very high in calcipenic variant as we can see in case no 1 where Parathyroid hormone and 1,25 OH vit D levels are very high with profound hypocalcemia. One of the striking features of VDDR type 2 is alopecia which occurs when there is mutation in DNA binding domain. These group of patients are refractory to conventional treatment with oral calcium. Some case reports showed that very high dose of oral calcium can cause significant biochemical and radiological improvement [1]. They may show spontaneous resolution at puberty and in some cases hypocalcemia can be so severe that regular IV calcium therapy may be needed. V kumar et al showed a case report where conventional dose of calcium with 1 a hydroxycholecalciferol improves the rachitic changes as well as alopecia in 4-year-old boy [2]. Another cause of refractory calcipenic rickets is 1a hydroxylase deficiency (VDDR 1A) where there is failure of formation of 1,25 OH vit D due to inactivating mutation of cyp27B1. There is a good genotype–phenotype correlation in VDDR-IA. However, some patients may recover from the loss of CYP27B1 function, probably due to 1a-hydroxylase activity exerted by a non-CYP27B1 enzyme [3]. If the initial suspicion goes in favor of phosphopenic ricket (dental abnormality, predominant lower limb involvement), TMP GFR has to be calculated to document presence of renal phosphate wasting and FGF23 has to be done to classify whether it is FGF23 dependent or independent. Low TMP GFR in background of phosphopenic rickets suggests renal phosphate wasting. Elevated or nonsuppressed FGF 23 in this background suggests FGF 23 dependent hypophosphatemic rickets among which XLH is the most common one which occurs due to inactivating PHEX gene mutation. Treatment options in XLH patients include oral phosphate and calcitriol supplementation to compensate renal phosphate wasting although recently Burosumab, a monoclonal antibody against FGF23 has been approved for management of these group of patients [4]. Others entities in this group are Autosomal Dominant Hypophosphatemic Rickets (ADHR), Autosomal Recessive Hypophosphatemic Rickets (ARHR). If FGF 23 level is low in a background of Low TMPGFR we have to suspect FGF23 independent rickets. In that case we have to look for disorders of renal tubular acidification collectively known as renal tubular acidosis. Proximal renal tubular acidosis commonly presents with generalized loss of calcium, phosphate, bicarbonate, glucose, amino acid, low molecular weight protein collectively known as Fanconi syndrome. These group of patients commonly presents with polyuria, polydipsia, rachitic changes, nephrocalcinosis, nephrolithiasis, growth failure like case no 3. Similar case series of rickets caused by proximal RTA had been published from our institute [5]. Except for RTA, other causes of FGF23 independent hypophosphatemia are also there like HHRH, presented in case no 5. In this group of patient’s calcitriol supplementation is not at all required with phosphate. As a whole refractory rickets require systemic evaluation to reach the final diagnosis and strict management and periodic follow up to prevent future morbidity and surgical correction.
References
- Wong GW, Leung SS, Law WY, Cheung NK, Oppenheimer SJ. Oral calcium treatment in vitamin D-dependent rickets type II. Journal of paediatrics and child health. 1994; 30: 444-6.
- Kumar V, Kumar M, Yadav M. Alopecia in vitamin D-dependent rickets type II responding to 1 a-hydroxycholecalciferol. Annals of tropical paediatrics. 2010; 30: 329-33.
- Durmaz E, Zou M, Al-Rijjal RA, Bircan I, Akçurin S, Meyer B, et al. Clinical and genetic analysis of patients with vitamin D-dependent rickets type 1 A. Clinical endocrinology. 2012; 77: 363-9.
- Lambert AS, Zhukouskaya V, Rothenbuhler A, Linglart A. X-linked hypophosphatemia: Management and treatment prospects. Joint Bone Spine. 2019; 86: 731-8.
- Singhania P, Dhar A, Deshpande A, Das D, Agrawal N, Chakraborty PP, et al. Rickets in proximal renal tubular acidosis: a case series of six distinct etiologies. Journal of Pediatric Endocrinology and Metabolism. 2023; 36: 879-85.